Etiology
Several genes have been identified in autosomal-dominant PKD (ADPKD). The most commonly affected genes are PKD1 or PKD2, with minor loci (e.g., GANAB, DNAJB11) accounting for a small proportion of often atypical patients, plus a group of genetically unresolved cases.[2][4][16]
PKD1 (chromosome region 16p13.3) is found in around 78% of cases and encodes the protein polycystin 1.[16]
PKD2 (4q21) is found in around 15% of cases and encodes polycystin 2.[16]
GANAB, a coding gene that encodes for a glucosidase-II alpha subunit, may account for approximately 0.3% of ADPKD cases.[16] However, due to the mild phenotype (nonenlarged cystic kidneys that often evolve to kidney atrophy) seen in individuals with mutations in GANAB, it is possible that this accounts for a larger proportion of the missing genetic causes of ADPKD.[17]
Mutations in the DNAJB11 gene, which encodes a glycoprotein of the endoplasmic reticulum (ER) required for ER homeostasis, are a rare (approximately 0.1% of ADPKD cases) cause of atypical ADPKD.[16] Cases have normal-sized cystic kidneys and progressive interstitial fibrosis resulting in late-onset end-stage renal disease.[18]
Combination of an ADPKD allele with an allele of another cystogene (e.g., both PKD1 and HNF1B) may present with an ADPKD-like phenotype.[1][19]
A family history of ADPKD is not apparent in 10% to 25% of patients with ADPKD.[20] This may be due to missing parental medical records, mild disease from hypomorphic PKD1 mutations (failure to recognize the disorder or late-onset disease in the affected parent), early death of the parent before the onset of symptoms, de novo mutations, or germline or somatic mosaicism.[16][20] Despite comprehensive screening, 10% to 15% of patients suspicious of ADPKD have no mutation detected in either PKD1 or PKD2.[21] Patients can be rescreened for mutations of a newly identified gene for ADPKD, GANAB, and somatic mosaicism.[20]
Pathophysiology
Patients with autosomal-dominant PKD (ADPKD) usually carry a germline mutation in one allele of either PKD1 or PKD2; however, there is notable phenotypic variability among family members who share a common mutation, suggesting that a “second hit” of a somatic mutation of the previously normal PKD1 allele plays a major role in determining the course of disease.[1][22] The theory is that a wild-type copy of the PKD1 gene develops an inactivating somatic mutation in a minority of cells, leading to loss of polycystin function and clonal cyst development.[22] The protein products of PKD1 and PKD2, polycystin 1 and polycystin 2 are membrane proteins that form a functional complex.[23] Evidence points to a role for the polycystins in the renal tubule primary cilium acting as a mechanosensitive cation channel, such that loss or dysfunction of polycystin 1 or polycystin 2 changes intracellular calcium concentration, which may then alter various cell functions, including gene expression, growth, differentiation, and apoptosis, thus altering tissue and organ development.[24] However, the biologic function of polycystins remains poorly understood.[25]
Renal cysts develop and grow over time, leading to compression of the normal renal architecture and intrarenal vasculature with obstructed nephrons forming atubular glomeruli and apoptotic proximal tubules, increased renal size, interstitial fibrosis, and progressive renal impairment.[1] Cysts develop from cells in the tubular portion of the nephron and collecting ducts. Although all cells are programmed with an ADPKD mutation, only a minority develop cysts. Kidneys progressively enlarge and become distorted with little recognizable parenchyma on imaging studies. Increasing combined renal volume is associated with decreasing renal function.[26] The average rate of decline of renal function is 4.4 to 5.9 mL/minute/year.[27][Figure caption and citation for the preceding image starts]: Gross pathology of polycystic kidneysAdapted from Dr Edwin P. Ewing, Jr., Public Health Image Library, CDC (1972) [Citation ends].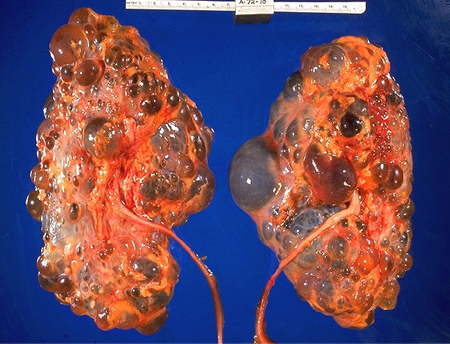
Based on findings of the landmark Modification of Diet in Renal Disease study, ADPKD patients had the most rapid decline of renal function of all forms of chronic kidney disease.[28] Individuals with PKD1 mutations have more severe disease with larger kidneys and earlier onset of end-stage renal disease (ESRD) compared with patients with PKD2 mutations.[2] Mean age of onset of ESRD is 55 years with a PKD1 mutation and 79 years with PKD2.[3] There are fewer kidney cysts in individuals with PKD2 than in those with PKD1, although the rate of growth of the kidneys does not differ between the groups.[29] PKD2-related ADPKD is typically milder than PKD1-related ADPKD; it should not be considered asymptomatic, and early referral, timely symptomatic care, and patient education may reduce environmental modulations of the phenotype.[30]
Additional disease-specific complications and morbidity arise from hepatic involvement, valvular heart disease, intracranial aneurysms, pain, massive kidney enlargement, and diverticular disease, contributing to additional morbidity and mortality.[1][31] PKD1 and PKD2 patients are equally likely to develop intracranial aneurysms, although patients with mutations in the 5' region of PKD1 are more likely to have aneurysm rupture.[32]
In autosomal-recessive PKD, renal cysts arise from collecting ducts, and congenital hepatic fibrosis is characteristic.[1][33]
Classification
Autosomal-dominant polycystic kidney disease (ADPKD)
ADPKD is a typically adult-onset disorder characterized by gradually growing renal cysts with progressive fibrocystic renal disease.[1] ADPKD is genetically and phenotypically heterogeneous. The most commonly affected genes are PKD1 or PKD2.
PKD1 (chromosome region 16p13.3) encodes the protein polycystin 1.
PKD2 (4q21) encodes polycystin 2.
Individuals with PKD1 mutations have more severe disease with larger kidneys and earlier onset of end-stage renal disease (ESRD) compared with patients with PKD2 mutations.[2] Mean age of onset of ESRD is 55 years with a PKD1 mutation and 79 years with PKD2.[3]
ADPKD is a systemic disease and extrarenal manifestations, such as liver cysts and cardiovascular abnormalities (including intracranial aneurysms), are present in both PKD1 and PKD2 variants.[1][4]
Autosomal-recessive polycystic kidney disease (ARPKD)
ARPKD is a rarer and often more severe form of cystic disease, which usually presents in children and is phenotypically highly variable, involving the kidneys and biliary tract.[1][5] Variants in the polycystic kidney and hepatic disease 1 (PKHD1) gene account for most cases of ARPKD, with mutations in DZIP1L a considerably rarer cause.[1]
At its most severe, it presents in utero or in the neonatal period with bilaterally enlarged echogenic kidneys and can be life-threatening.[1][5] In older children it manifests as portal hypertension or cholangitis.[1]
Early-onset, severe phenotype ADPKD may appear to be clinically indistinguishable from ARPKD or, alternatively, an advancing clinical course of ARPKD may resemble the symptoms observed in ADPKD; however, in contrast to the persistent renal enlargement that occurs in ADPKD, in ARPKD the kidneys decrease in size as the amount of fibrosis increases.[1][5] Next-generation sequencing-based screening of a panel of targeted genes is considered the most efficient approach to diagnosis.[1]
This topic does not deal with ARPKD.
Use of this content is subject to our disclaimer