Approach
Patients with classic CF will generally present in infancy or early childhood with failure to thrive. Some infants and young children will present with prolonged or severe bronchiolitis or recurrent respiratory complaints. Undiagnosed adults with CF, who are usually pancreatic sufficient, may present with chronic or recurrent bronchitis, rhinosinusitis, or pancreatitis.
CF is diagnosed when the individual has both a clinical presentation of CF and evidence of CFTR dysfunction.[28] Most patients in the US are diagnosed immediately after birth due to newborn screening.
History
The birth history should include questions about the passage of meconium (any delay) and the place of birth (US hospitals perform newborn screening). A positive or suspicious family history raises the level of suspicion for CF.
Respiratory tract
Question the family about the respiratory rate, retractions, cough (quantity and quality, including sputum production), and wheeze. A wet-sounding cough, particularly with hard coughing spells, may indicate CF.
Patients may present with recurrent lower airways infections (bronchitis or pneumonia) that require antibiotics. Pulmonary exacerbations often present with blood-streaked sputum, but some patients may experience major hemoptysis (>300 cc/24 hours).
Gastrointestinal tract
Question patients or parents about appetite, typical foods eaten, stooling habits (including quantity and quality), and any gastroesophageal reflux.
An insatiable appetite, with frequent bulky and greasy stools, is consistent with fat and calorie malabsorption.
A history of decreasing stool numbers over time, with or without abdominal distention or vomiting, may signal bowel obstruction. This is more common in people with CF than in those without.
Suspect exocrine pancreatic insufficiency in patients with CF. Usually presents at birth or in infancy.[29]
Patients may present with recurrent pancreatitis and/or acute appendicitis. Having the CFTR selective bicarbonate conductance defect (except p.R75Q) increases the risk of chronic pancreatitis two- to fourfold.[30]
Physical exam
The physical exam may be normal in a patient with CF. However, a few key findings should raise the level of suspicion.
Malnutrition, manifest as a lack of subcutaneous fat stores, protuberant abdomen, and below normal weight-for-height in infants and young children. BMI in older children, adolescents, and adults.
Nasal polyps. See Nasal polyps.
Increased anteroposterior diameter of the chest, and crackles at auscultation.
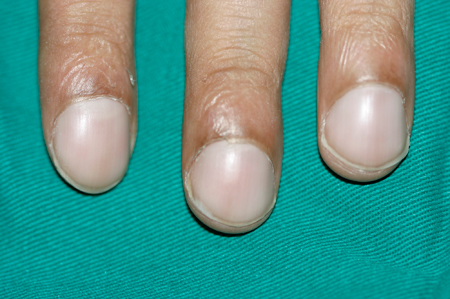
Digital clubbing of the hands. See Assessment of clubbing.
Stool mass (often in the right lower quadrant), or an enlarged liver and/or spleen, or both. Revealed by abdominal palpation.
Bilateral absence of the vas deferens, in males.[Figure caption and citation for the preceding image starts]: Finger clubbingFrom the collection of Dr Murlidhar Rajagopalan [Citation ends].
 [Figure caption and citation for the preceding image starts]: CT—Coronal view of the paranasal sinuses showing the blockage of the osteomeatal complexes (Green Circle)Mohd Slim MA et al. Paediatric nasal polyps in cystic fibrosis. BMJ Case Rep. 2016 Jun 21;2016; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: CT—Coronal view of the paranasal sinuses showing the blockage of the osteomeatal complexes (Green Circle)Mohd Slim MA et al. Paediatric nasal polyps in cystic fibrosis. BMJ Case Rep. 2016 Jun 21;2016; used with permission [Citation ends]. [Figure caption and citation for the preceding image starts]: Pale-color soft tissue mass in the right nasal cavityMohd Slim MA et al. Paediatric nasal polyps in cystic fibrosis. BMJ Case Rep. 2016 Jun 21;2016; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Pale-color soft tissue mass in the right nasal cavityMohd Slim MA et al. Paediatric nasal polyps in cystic fibrosis. BMJ Case Rep. 2016 Jun 21;2016; used with permission [Citation ends].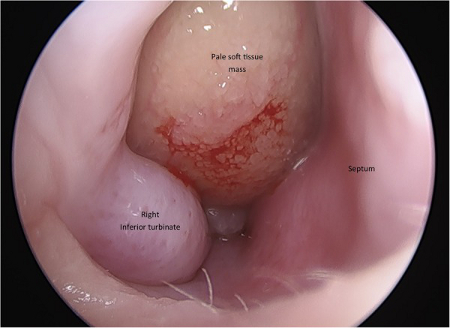
Investigations
Cystic Fibrosis Foundation guidance recommends that tests should ideally be performed hierarchically to establish the diagnosis: sweat tests first, followed by CFTR genetic tests, and then CFTR physiologic tests. All people diagnosed with CF should have at least a sweat test and a CFTR genetic analysis performed.
Sweat test
If suspected (i.e., patient presents with symptoms/signs or a positive family history), a sweat test (pilocarpine iontophoresis test) should be performed.
Negative sweat test: sweat chloride measurement of <30 mEq/L (in all age groups) suggests that CF is unlikely.[28] However, if questions remain, referral to a CF center is recommended even in the presence of a negative sweat test.
Positive sweat test: sweat chloride measurement of ≥60 mEq/L is consistent with CF and requires immediate referral to a CF center.
Intermediate sweat test: sweat chloride measurement of 30-59 mEq/L; CFTR genetic analysis is required.
Sweat tests may be performed in children of any age. Some children may not produce enough sweat to give accurate results. If this occurs, the child should be retested within a week.
CFTR genetic tests
Genetic testing can help to establish the diagnosis. Most laboratories will perform an initial "screen" for the most common cystic fibrosis transmembrane conductance regulator (CFTR) mutations. If two common mutations are not found, most laboratories have an option for sequencing more of the CFTR gene or the entire CFTR gene.
In individuals with sweat test results that fall within the intermediate range (defined as a sweat chloride measurement of 30-59 mEq/L), it remains crucial to evaluate further with a CFTR genetic analysis. Subsets of these patients can be characterized as having CFTR-related metabolic syndrome (CRMS), also known as a CF screen-positive, inconclusive diagnosis (CFSPID) in Europe.
If there are existing genetic test results, do not order a duplicate test unless there is uncertainty about the existing result, e.g., the result is inconsistent with the patient’s clinical presentation or the test methodology has changed.[31]
Physiologic and ancillary tests
Physiologic tests include nasal potential difference and intestinal current measurement. Ancillary tests include sinus imaging, which may show pansinusitis, and a deep throat swab, which may demonstrate respiratory pathogens.[32] Neither sinus imaging or throat swab are specific tests for CF.
In CF patients presenting with appendicitis, the pathologic finding of inspissated, hematoxylin-staining material within the crypts of the appendix is pathognomonic.[33]
Newborn screening
The Cystic Fibrosis Foundation recommends appropriately performed newborn screening for all infants.[34] Although all states in the US include CF in their newborn screening panels, this should not replace prepregnancy or prenatal carrier screening.[35]
Use of this content is subject to our disclaimer