Approach
This is a challenging disorder to diagnose, categorize, and treat due to its varied presentation and uncertain course. The integral diagnostic test for pityriasis lichenoides is histology. Histology can also delineate different subtypes of pityriasis lichenoides.[4] Dermoscopy can differentiate between pityriasis lichenoides chronica and guttate psoriasis, decreasing the number of cases requiring biopsy.[18]
If infectious etiology is suspected, evaluation of antistreptolysin O titers, throat cultures, Epstein-Barr virus antibodies, monospot or heterophile antibodies, toxoplasma Sabin-Feldman dye, and serological diagnosis of toxoplasmosis, hepatitis, and HIV are recommended.[4][6]
Clinical evaluation
Pityriasis lichenoides et varioliformis acuta, also known as Mucha-Habermann disease, presents as acute eruption of numerous, small, erythematous papules that rapidly develop into polymorphic lesions at various stages of development, such as vesicles, pustules, hemorrhagic crust-covered papules, and shallow ulcers.[3] The lesions show predilection for the anterior torso, flexural surfaces, and proximal extremities, although they may appear in other areas. Rarely, the lesions may be associated with burning, pruritus, and mild constitutional symptoms.[1] Low-grade fever, lymphadenopathy, malaise, myalgias, arthralgias, and headache may precede or appear simultaneously with the eruption signaling a potential infectious source.
Pityriasis lichenoides chronica manifests more gradually as small, pink-to-brown macules and papules with thin, centrally attached scales.[3] The crops of these lesions are usually scattered across the torso and proximal extremities.
The febrile ulceronecrotic Mucha-Habermann disease variant is a very rare and severe form of pityriasis lichenoides that presents as sudden and generalized eruption of purpuric-black, ulceronecrotic and crusted plaques, which may involve mucosal areas.[3][19] Lesional pain, pruritus, high fever, constitutional symptoms, and multiorgan injury are the other prominent presentations.[1][Figure caption and citation for the preceding image starts]: Pink-to-red necrotic, excoriated, crusted papules scattered over a child's back and buttocks is a presentation of pityriasis lichenoides et varioliformis acutaFrom the collection of Dr A. Khachemoune, SUNY Downstate Medical Center, Brooklyn. [Citation ends]. [Figure caption and citation for the preceding image starts]: Closer view of the pityriasis lichenoides chronica papules with micaceous scaleFrom the collection of Dr A. Khachemoune, SUNY Downstate Medical Center, Brooklyn. [Citation ends].
[Figure caption and citation for the preceding image starts]: Closer view of the pityriasis lichenoides chronica papules with micaceous scaleFrom the collection of Dr A. Khachemoune, SUNY Downstate Medical Center, Brooklyn. [Citation ends].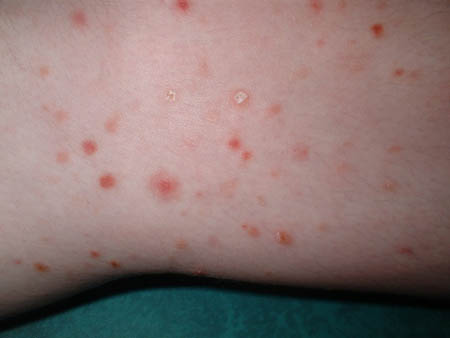 [Figure caption and citation for the preceding image starts]: Pityriasis lichenoides chronica presenting as pink-to-light-brown small, flat papules on a child's armsFrom the collection of Dr A. Khachemoune, SUNY Downstate Medical Center, Brooklyn. [Citation ends].
[Figure caption and citation for the preceding image starts]: Pityriasis lichenoides chronica presenting as pink-to-light-brown small, flat papules on a child's armsFrom the collection of Dr A. Khachemoune, SUNY Downstate Medical Center, Brooklyn. [Citation ends].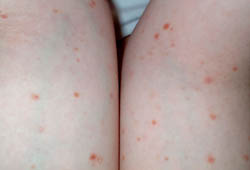
Risk factors
Pityriasis lichenoides tends to present in late childhood and early adulthood, and shows a slight predisposition in males. Patients with HIV also have a slight predisposition for developing the condition.
Histopathologic exam
Histopathologic examination confirms the diagnosis of pityriasis lichenoides. Pityriasis lichenoides et varioliformis acuta is characterized by wedge-shaped, dense, and diffuse dermal CD8+ predominant lymphocytic infiltrate concentrated along the basal layer with prominent lymphocytic exocytosis into the epidermis.[20] Next-generation sequencing for T-cell clonality may be positive, although it is not specific for pityriasis lichenoides.[20][21] Routine performance of T-cell receptor gene rearrangement is not required when there is clinical and histologic concordance for a diagnosis of pityriasis lichenoides et varioliformis acuta or lymphomatoid papulosis.[21][22] The epidermis shows some apoptotic and necrotic keratinocytes, neutrophilic inclusions, spongiosis, and focal parakeratosis. The histopathology of pityriasis lichenoides chronica reveals a superficial CD4+ T-dominant lymphocytic, band-like, sparsely perivascular infiltrate. It also exhibits slight parakeratotic scale, minimal keratocytic necrosis, and spongiosis in the epidermis. The febrile ulceronecrotic Mucha-Habermann disease variant demonstrates a more prominent lymphocytic and neutrophilic infiltrate, pronounced epidermal necrosis and ulceration, and leukocytoclastic vasculitis.[19][20]
Serologic studies
If an infectious pathogen is suspected, blood serologies for associated agents may be checked to assist with causal investigation. In a small series, 79% of patients had a positive antistreptolysin titer.[6] Febrile ulceronecrotic Mucha-Habermann disease may demonstrate multiple laboratory abnormalities, such as leukocytosis, hypoalbuminemia, serum elevation of erythrocyte sedimentation rate, C-reactive protein, and lactic dehydrogenase, which are markers of systemic inflammation and help to solidify the diagnosis.[1][23]
Use of this content is subject to our disclaimer