Etiology
The etiology of Takayasu arteritis is unknown. Environmental and genetic factors are thought to play roles in the development of the disease.[4] Cell-mediated immune mechanisms have been implicated.[1] Genetic screening has shown polymorphisms in IL-12, IL-6, and IL-2 genes in a population of Turkish patients with Takayasu arteritis.[15] HLA-Bw5 and HLA-B39.2 are reportedly increased in frequency in some populations.[16][17]
Pathophysiology
Takayasu arteritis is an immune-mediated vasculitis characterized by granulomatous inflammation of large arteries. [Figure caption and citation for the preceding image starts]: Photomicrograph of the aorta from a patient with Takayasu arteritis demonstrates marked thickening of the intimal layer and inflammatory infiltrates in the media and laminar necrosisUsed with permission from the collection of Dylan Miller, MD, Mayo Clinic [Citation ends].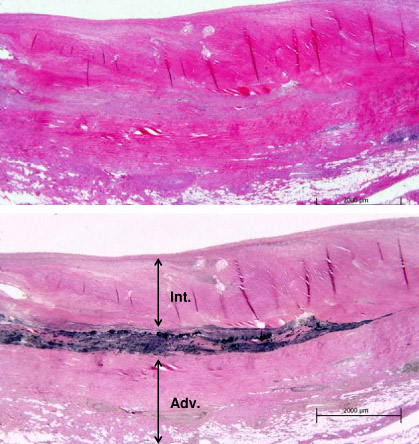 Cell-mediated immune mechanisms have been implicated.[1][4] Interleukin (IL)-6 and IL-17 are thought to play an important role in the pathogenesis of Takayasu arteritis.[18] Some patients have been treated with an IL-6 inhibitor with favorable responses.[19][20]
Cell-mediated immune mechanisms have been implicated.[1][4] Interleukin (IL)-6 and IL-17 are thought to play an important role in the pathogenesis of Takayasu arteritis.[18] Some patients have been treated with an IL-6 inhibitor with favorable responses.[19][20]
The immunologic and inflammatory response seen in arteries is similar to that observed in large arteries in giant cell arteritis.[1][4] During the acute phase of vasculitis, inflammation begins in the vasa vasora of the adventitia of muscular arteries.[1][9] T cells are prominent in the initial cellular response, and antiendothelial cell antibodies may also be involved.[1][4][21][22]
Classification
2012 International Chapel Hill Consensus Conference on the Nomenclature of Vasculitides[5]
Categorizes vasculitis based upon the predominant type of vessels involved, and other features including etiology, pathogenesis, type of inflammation, favored organ distribution, clinical manifestations, genetic predispositions, and distinctive demographic characteristics.
Large vessel vasculitis
Takayasu arteritis
Giant cell arteritis
Medium vessel vasculitis
Polyarteritis nodosa
Kawasaki disease
Small vessel vasculitis
ANCA-associated vasculitis
Microscopic polyangiitis
Granulomatosis with polyangiitis (formerly known as Wegener granulomatosis)
Eosinophilic granulomatosis with polyangiitis (Churg-Strauss)
Immune complex vasculitis
Anti-glomerular basement membrane (anti-GBM) disease
Cryoglobulinemic vasculitis
IgA vasculitis (Henoch-Schönlein)
Hypocomplementemic urticarial vasculitis
Variable vessel vasculitis
Behçet disease
Cogan syndrome
Single-organ vasculitis
Vasculitis associated with systemic disease
Vasculitis associated with probable etiology
Angiographic classification of Takayasu arteritis[6]
Classification is based on the vessels involved in the inflammatory process as seen on angiography.
Type I: Branches of the aortic arch
Type IIa: Ascending aorta, aortic arch, and branches of the aortic arch
Type IIb: Ascending aorta, aortic arch, and its branches and thoracic descending aorta
Type III: Thoracic descending aorta, abdominal aorta, and/or renal arteries
Type IV: Abdominal aorta and/or renal arteries
Type V: Features of types IIb and IV
Use of this content is subject to our disclaimer