Approach
Most clinically nonfunctional pituitary adenomas (CNFPAs) present late because they either do not secrete any hormones or they secrete functional hormones inefficiently.[1] Mean age of presentation is 40 to 58 years for women versus 58 to 62 years for men.[34]
History
Pituitary adenomas may be found incidentally on imaging, or may present with symptoms and signs of mass effect or neuroendocrine changes. Headaches are common (35% to 70%) but the underlying pathophysiology remains uncertain.[35] Possible mechanisms include structural causes such as dural stretch or cavernous sinus invasion.[8] Tumor growth into the third ventricle may cause hydrocephalus and associated symptoms such as headache, imbalance, and urinary incontinence.[8] Lateral tumor growth into the cavernous sinus may cause double vision from third, fourth, and sixth cranial nerve palsies and facial pain and paresthesia from palsy of the V1 and V2 branches of the fifth cranial nerve.[9] Seizures may occur from temporal lobe involvement.[10] Recurrent sinusitis and cerebrospinal fluid rhinorrhea may result from growth into the sphenoid sinus.
Patients with CNFPAs may present with features of hypopituitarism.[8][36] The gonadotrophs and somatotrophs appear most susceptible to the local pressure damage from a pituitary adenoma. Hypogonadism in men presents with loss of secondary sexual characteristics, mood impairment, loss of libido, erectile dysfunction, infertility, anemia, loss of muscle mass, and osteopenia. In women, amenorrhea, diminished libido, infertility, hot flashes, osteopenia, and breast atrophy are present. Growth hormone deficiency causes truncal obesity; fatigue associated with diminished effort tolerance, diminished muscle mass and increased fat mass, osteopenia, depression, and abnormal lipid profile. Hypothyroidism causes fatigue, constipation, weight gain, cold intolerance, dry skin, hair loss, bradycardia, memory loss, and depression. Adrenal insufficiency causes fatigue, nausea, anorexia, weight loss, hyponatremia, weakness, and trembling. There is typically no hyperkalemia because the mineralocorticoid pathway is intact and there is no hyperpigmentation.
Gonadotroph adenomas are usually nonfunctional, but rarely present with clinical findings secondary to hypersecretion of gonadotrophins, including precocious puberty in children, macro-orchidism in males, ovarian hyperstimulation in premenopausal women who have follicle-stimulating hormone-secreting tumors, or abnormally high testosterone levels with luteinising hormone-secreting tumors.[1][14]
Diabetes insipidus is a rare finding at diagnosis of CNFPAs, although it may result from surgery of the pituitary adenoma.[37]
Pituitary apoplexy
May be the presenting clinical picture with severe headache of sudden onset, fever, nausea and vomiting, meningismus, altered level of consciousness, visual disturbances, and hypopituitarism.[11] Clinically significant pituitary apoplexy is a rare event in patients with pituitary microadenomas. The risk of apoplexy is estimated to be from 2% to 12% of patients with pituitary adenomas.[11] It is due to a rapid enlargement of the pituitary as a result of either hemorrhage and/or infarction of the tumor.[12] While most cases of pituitary apoplexy are spontaneous, precipitating factors may include head injury, anticoagulant therapy, dopamine agonists, radiation therapy, or dynamic endocrine tests. In a meta-analysis of patients with pituitary incidentalomas and CNFPAs, pituitary apoplexy occurred at a rate of 0.2 per 100 person-years, with a non statistically significant trend for a higher rate of apoplexy with macroadenomas compared with microadenomas.[38]
If the hemorrhage or infarction of the tumor is found to have happened at some point in the past, the patient may recall no symptoms, and can be considered as having had asymptomatic pituitary apoplexy.
Exam findings
At presentation 80% have visual field defects as a result of visual pathway compression, manifesting most commonly as bitemporal hemianopia with chiasmal compression.[37] Patients may have diminished visual acuity; visual field loss typically starts with bitemporal superior quadrantanopia.[39] Cranial nerve palsies involving third, fourth, fifth (V1 and V2), and sixth cranial nerves and mydriasis associated with third nerve palsy may be present. Patients with pituitary apoplexy may have variable degrees of change in mental status.
Endocrine-related findings in patients with pituitary adenoma include: signs of hypogonadism (decreased facial and body hair, gynecomastia, decreased muscle mass, soft testicles in men and breast atrophy in women); hypothyroidism (dry skin, coarse hair, puffy face, loss or thinning of eyebrows); and growth hormone deficiency (loss of muscle mass, increased abdominal obesity). Pallor and increased skin wrinkling are characteristic findings in patients with panhypopituitarism.
Imaging
A dedicated pituitary MRI pre- and post-gadolinium enhancement is preferred over a CT scan.[39][40] The MRI will delineate the tumor characteristics, the presence of tumor invasion into cavernous sinuses and the sphenoid sinus, and any mass effect on optic chiasm.[Figure caption and citation for the preceding image starts]: Precontrast coronal (A) and sagittal (B) MR images of a patient with a pituitary macroadenoma (arrow, A). The pituitary mass extends toward the optic chiasm with some pressure effectFrom the collection of Dr Amir Hamrahian [Citation ends].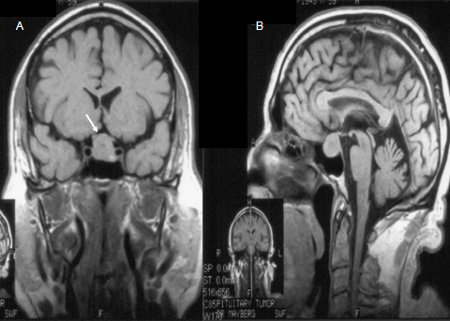 [Figure caption and citation for the preceding image starts]: Precontrast (A) and postcontrast (B) coronal MR images of a patient with a small pituitary microadenoma. The pituitary lesion enhances less than the normal pituitary gland following gadolinium and appears hypodense compared with the normal pituitary gland (arrow, B)From the collection of Dr Amir Hamrahian [Citation ends].
[Figure caption and citation for the preceding image starts]: Precontrast (A) and postcontrast (B) coronal MR images of a patient with a small pituitary microadenoma. The pituitary lesion enhances less than the normal pituitary gland following gadolinium and appears hypodense compared with the normal pituitary gland (arrow, B)From the collection of Dr Amir Hamrahian [Citation ends].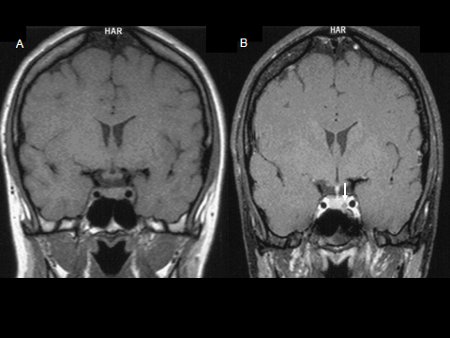
Ophthalmic evaluation
Eye examination and formal visual field testing (Humphrey or Goldmann visual field test) are indicated if imaging shows the adenoma is causing pressure on, or has contact with, the optic chiasm, in order to document visual acuity and field deficits.
Routine blood tests
In addition to the hormonal evaluation, a basic metabolic panel and CBC should be done initially. Hyponatremia may be seen in adrenal insufficiency and hypothyroidism and is also associated with symptoms such as nausea, vomiting, headache, and malaise. Anemia may be a feature of patients with longstanding hypogonadism, hypothyroidism, or adrenal insufficiency and is associated with fatigue and malaise. A lipid panel may be done subsequently to determine hyperlipidemia and risk of premature atherosclerosis in growth hormone deficiency and hypogonadism.
Prolactin
Prolactin levels associated with prolactinomas correspond with tumor size. Mild to moderate hyperprolactinemia may be present (<100 nanograms/mL) with a micro-prolactinoma, or, in the presence of a pituitary macro-adenoma, is usually due to pituitary stalk compression.[41] With the exception of patients on medications such as metoclopramide and antipsychotics, prolactin level >200 nanograms/mL is almost always diagnostic of a prolactinoma. Other physiological causes of hyperprolactinemia include pregnancy, chest wall or nipple stimulation, renal failure, hypothyroidism and adrenal insufficiency.
Growth hormone and insulin-like growth factor 1
Growth hormone (GH) deficiency is one of the most common hormone deficiencies associated with CNFPAs. Random GH levels are not helpful in making the diagnosis and insulin-like growth factor 1 (IGF-1) may be normal in up to 65% of patients with GH deficiency, and is also not regarded as an adequate screening test. The presence of three or more anterior pituitary hormone deficiencies in the presence of low IGF-1 almost always indicates the presence of GH deficiency and thus may obviate the need for further testing.[42] However, a stimulated GH level with either an insulin tolerance test (ITT), glucagon stimulation test, or arginine/growth hormone-releasing hormone (GHRH) stimulation test, or macimorelin test, is typically required for the diagnosis. GHRH is currently only available on special request in the US. While ITT is the diagnostic standard, it needs to be performed by an experienced endocrinologist, because of the risks associated with iatrogenic hypoglycemia and the need to administer glucose for recovery when necessary. ITT is contraindicated in patients with history of seizure disorder, coronary artery and cerebrovascular disease, and in those older than 65 years. The glucagon stimulation test has been proposed as an alternative to ITT for evaluation of patients suspected to have GH deficiency.[43] Macimorelin is an oral GH secretagogue receptor agonist that has been approved for the diagnosis of GH deficiency in adults.[44]
Follicle-stimulating hormone, luteinising hormone, alpha subunits of human pituitary glycoprotein hormones, estradiol, and testosterone
Low serum testosterone in men, or estradiol in women, accompanied by normal/low follicle-stimulating hormone (FSH) and luteinising hormone (LH) levels are consistent with gonadotropin deficiency in males and amenorrheic premenopausal women. Failure to elevate FSH and LH in postmenopausal women is also consistent with gonadotropin deficiency. The presence of regular menses almost always indicates a normal gonadotropin axis. In women with irregular menstruation, the interpretation of hormonal evaluation for gonadotropin axis can be challenging and usually is not indicated.
Although over 45% of CNFPAs are in fact gonadotrope cell adenomas, they seldom produce a hormonal hypersecretion syndrome.[37] They can produce intact FSH and LH, but more commonly produce varying combinations of either FSH beta subunit and/or LH beta subunit along with the common alpha subunit.[37] In those with gonadotroph cell adenomas, FSH, LH, and/or alpha subunit may be elevated. Rarely, testosterone may be elevated in those with LH-producing pituitary adenomas.
Thyroid-stimulating hormone and free or total thyroxine
If secondary hypothyroidism is clinically suspected, thyroid-stimulating hormone (TSH) and free T4 (or free T4 index) should be measured together. Typically patients have a low or, more commonly, normal TSH along with a low free T4 level, in contrast to patients with primary hypothyroidism, in whom TSH is elevated.
Adrenocorticotropic hormone and cortisol
Adrenocorticotropic hormone (cosyntropin) stimulation test and early morning (e.g., 8 am) plasma cortisol level are reasonable initial tests for evaluation of the corticotropin axis.
Early morning cortisol level less than 3 micrograms/dL strongly suggests adrenal insufficiency; while a value greater than 15 micrograms/dL makes the diagnosis highly unlikely.
Cortisol levels in the range of 3-15 micrograms/dL are indeterminate and should be further evaluated by adrenocorticotropic hormone (ACTH) stimulation test, which can be performed any time during the day.
The optimal cortisol values used in the ACTH stimulation test need to be individualized depending on the assay used, as newer assays (e.g., monoclonal antibody immunoassays or mass spectrometry) may have a lower threshold and result in a false positive test.[45] The value of <18 micrograms/dL to indicate adrenal insufficiency is derived from polyclonal antibody assays; a lower serum cortisol cutoff of less than 14-15 micrograms/dL is suggested for the new specific monoclonal antibody immunoassays.[45]
Cut-off values also need to be individualized according to whether high-dose or low-dose cosyntropin is used for the ACTH stimulation test.[46] The standard dose ACTH stimulation test utilizes an intravenous or intramuscular injection of 250 micrograms cosyntropin with plasma cortisol levels before, and then 30 minutes after, the injection. A normal response is a plasma cortisol concentration greater than 18 micrograms/dL at 30 minutes. Patients with mild, partial, or recent-onset pituitary ACTH or hypothalamic corticotropin-releasing hormone deficiency (e.g., within 2 to 4 weeks after pituitary surgery) may have a normal response to 250 micrograms cosyntropin because the adrenal glands have not undergone sufficient atrophy and still respond to very high concentrations of ACTH stimulation. The sensitivity of the ACTH stimulation test to identify mild, partial adrenal insufficiency improves with the use of low dose cosyntropin (1 microgram).[46]
ACTH levels are not reliable for diagnosis but can be used to differentiate primary and secondary adrenal insufficiency in those with low cortisol levels.
Gonadotropin-releasing hormone stimulation test
This may be useful in evaluation of the gonadotropin axis in selected pediatric patients, but it is generally not needed. There have been some reports about the test inducing pituitary apoplexy.
Immunohistochemical staining
Done after resection of the tumor, and it may help to shape diagnosis and treatment based on subclinical secretion of hormones.
Use of this content is subject to our disclaimer