Aetiology
Focal segmental glomerulosclerosis (FSGS) is initiated by an injury to podocytes in the renal glomeruli. In primary FSGS, this injury is produced by, as yet, unidentified circulating factors that alter the permeability of the glomeruli.[22] Proposed candidate molecules include haemopexin, vascular endothelial growth factor, soluble form of urokinase receptor (suPAR), and cardiotrophin-like cytokine-1.
suPAR is one candidate molecule that has been extensively investigated. In one study, elevated levels of suPAR were present in the plasma of two-thirds of patients with FSGS.[23] One meta-analysis also demonstrated that suPAR levels were significantly higher in patients with primary FSGS compared to controls.[24]
Some forms of primary FSGS are inherited and a positive family history may suggest a genetic cause. Mutations in a range of podocyte proteins have been associated with FSGS.[25] The majority of the genetic causes in children follow an autosomal recessive pattern with mutations in the genes nephrin (NPHS1), podocin (NPHS2), and phospholipase C epsilon 1 (PLCE1) being the most common.[26] Autosomal dominant causes of FSGS are more common in adolescents and adults. Major genes implicated in autosomal dominant FSGS include alpha-actinin 4 (ACTN4), inverted formin 2 (INF2), and the transient receptor potential cation 6 channel (TRPC6).[25] MYO1E mutations have been associated with childhood-onset, glucocorticoid-resistant FSGS.[27]
In secondary FSGS, podocyte injury is produced by viruses, toxins, intrarenal haemodynamic changes, or a maladaptive response to the loss of functioning nephrons. However, it is unclear why the injury produced by these diverse causes specifically targets glomerular podocytes.
The underlying conditions that produce secondary FSGS include:
Viral infection: HIV is the most common cause. Rarer causes include persistent parvovirus B19 infection, cytomegalovirus, hepatitis B and C, and SARS-CoV-2.
Glomerular hyperfiltration due to reduced renal mass: renal mass may be reduced by having a solitary kidney (due to nephrectomy or unilateral renal agenesis), reflux nephropathy, renal dysplasia, or chronic allograft nephropathy (in renal transplant recipients).
Drug-induced: heroin, interferon alfa, lithium, pamidronate, and sirolimus are known causes.
Obesity.
Vascular: thromboembolism and renal ischaemia are rare causes.
Black patients have a higher risk of developing end-stage renal failure due to primary or secondary FSGS and tend to develop renal failure at a younger age than white or Asian patients. The increased propensity toward FSGS may be related to mutations in MYH9, which are more common in black patients. These mutations may cause primary FSGS and exacerbate HIV-induced FSGS. Apolipoprotein L1 (APOL1) gene polymorphisms are also associated with FSGS in black patients.[28] These appear to be expressed almost exclusively in individuals of African descent, but have less commonly been identified in other populations.[29]
Pathophysiology
Injury to podocytes triggers apoptosis, causing them to detach from the glomerular basement membrane and be destroyed.[1] As podocyte numbers decline, the glomerular basement membrane is exposed, and maladaptive interactions develop between the glomerular basement membrane and the parietal epithelial cells. This is followed by the proliferation of epithelial, endothelial, and mesangial cells. The combination of cell proliferation and leak of protein into Bowman's space results in the deposition of collagen. Ultimately, the capillary loop collapses and endothelial cells are lost.[30] The glomerular tuft undergoes sclerosis, creating the characteristic lesion of FSGS. The extent of the lesions initially varies between different regions of the kidney. However, the disease eventually progresses to produce global sclerosis and end-stage renal failure. [Figure caption and citation for the preceding image starts]: Characteristic features of focal segmental glomerulosclerosis (FSGS) lesionsCreated at the BMJ Knowledge Centre [Citation ends].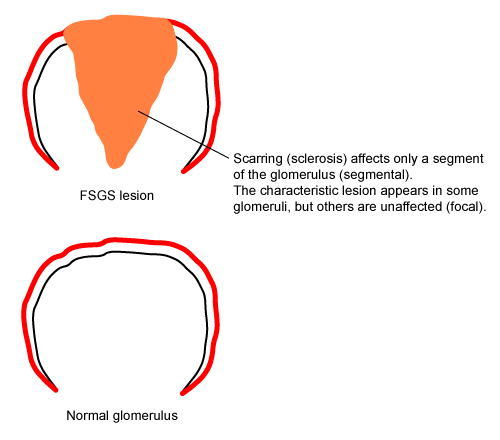
Once podocytes begin to die, protein leaks across the glomerular membrane, resulting in proteinuria and hypoalbuminaemia. At the same time, cholesterol levels rise due to increased cholesterol synthesis in the liver and the loss of lipid-regulating proteins in urine; the mechanism of these effects is incompletely understood.
The severity of FSGS differs between primary and secondary FSGS. Primary FSGS produces more severe podocyte injury and is most frequently associated with nephrotic syndrome. The injury produced by secondary FSGS is less severe, and proteinuria with hypoalbuminaemia in the nephrotic range is less common. This difference is reflected by the pattern of podocyte injury detected by electron microscopy. FSGS produces fusion of podocyte foot processes.[31] In primary FSGS, foot process fusion is diffuse and occurs throughout the glomeruli. In secondary FSGS, foot process fusion is mostly limited to the sclerotic areas.
Classification
Aetiological classification
Primary (idiopathic) FSGS
Mediated by unidentified circulating/permeability factors and characterised by diffuse foot process effacement and nephrotic syndrome.
Secondary FSGS
Caused by an acquired underlying condition.
Genetic FSGS
Develops in patients with mutations in genes encoding podocyte or glomerular basement membrane proteins.
FSGS of undetermined cause
Characterised by segmental foot process effacement and proteinuria without nephrotic syndrome, with no evidence of secondary causes.
Morphological classification[2][3]
Perihilar variant
At least 1 glomerulus has perihilar hyalinosis, with or without sclerosis.
>50% of glomeruli with segmental lesions must have perihilar sclerosis and/or hyalinosis.
Cellular variant
At least 1 glomerulus has segmental endocapillary hypercellularity occluding lumina, with or without foam cells and karyorrhexis.
Tip variant
At least 1 segmental lesion involves the tip domain (outer 25% of tuft next to origin of the proximal tubule).
Collapsing variant
At least 1 glomerulus has segmental or global collapse and overlying podocyte hyperplasia.
Some investigators believe that this is not a form of FSGS but a unique disorder called collapsing glomerulopathy.
Not otherwise specified variant
At least 1 glomerulus has segmental increase in matrix obliterating the capillary lumen.
Use of this content is subject to our disclaimer