Aetiology
While MS was classically viewed as a disease of central nervous system white matter, there is now substantial evidence supporting both grey and white matter involvement.[16] It appears to have both inflammatory and degenerative components that may be triggered by an environmental factor or factors in people who are genetically susceptible.
First-degree family members of patients with MS are 20-40 times more likely to develop MS than the general population.[17][18] The age-adjusted lifetime risk for children who have one parent with MS is about 2% to 3%.[19] The worldwide prevalence of familial MS has been estimated to be 12.6%.[20] While the genetics of MS are multifactorial, genes in the human leukocyte antigen (HLA) region and the interleukin region are likely to be involved.[21][22] Genetic testing to predict risk for MS is not currently recommended.
Environmental factors that have been postulated to be involved in MS include toxins, viral exposure, and sunlight exposure (and its effect on vitamin D metabolism).[23][24] While some researchers have proposed that MS is caused by a virus, none of the nearly 20 viruses that have been candidates over the last 20 years have proved to be causative. Epstein-Barr virus is the virus with the greatest link to increased risk of MS.[25][26] Human herpes virus 6 has also been suggested to be linked to MS but this has not been conclusively demonstrated.[27]
Relapses are sometimes triggered by infections or postnatal hormonal changes. Some literature suggests that acute trauma or stressful events may be precipitants, although this is controversial.[28][29]
Pathophysiology
The precise pathogenesis of MS is unknown. There is no specific or sensitive antigen or antibody, and there is some debate about whether MS represents a single disease or a syndrome of pathogenically heterogeneous patient subgroups. Conceptualisations of MS immunopathology involve 2 distinct but overlapping and connected phases, inflammatory and degenerative.
During the initial stage of the inflammatory phase, lymphocytes with encephalitogenic potential are activated in the periphery by factors such as infection or other metabolic stress. These activated T cells seek entry into the central nervous system (CNS) via attachment to a receptor on endothelial cells. This interaction, mediated by production of matrix metalloproteinases, allows a breach in the blood-brain barrier, leading to further upregulation of endothelial adhesion molecules and additional influx of inflammatory cells. The T cells produce inflammatory cytokines that cause direct toxicity and also attract macrophages that contribute to demyelination. Epitope spread occurs early and contributes to the complexity of the immunopathology.
The degenerative component of MS is believed to reflect axonal degeneration and loss. Demyelination disrupts axonal support and leads to destabilisation of axonal membrane potentials, which causes distal and retrograde degeneration over time. There is also a suggestion that inflammatory cells, antibodies, and complement may contribute to axonal injury. Axonal damage has been identified in regions of active inflammation, indicating that it begins early in the disease process.[30]
Pathologically, MS is characterised by multifocal areas of demyelination, loss of oligodendrocytes, and astrogliosis with loss of axons primarily in the white matter of the CNS, although cortical lesions may also play a significant role. Clinical heterogeneity and studies of biopsy and autopsy specimens suggest that the mechanisms leading to tissue damage differ from patient to patient.
Relapsing-remitting MS shows the most inflammatory activity, followed by early secondary progressive MS. Primary progressive MS is thought to be a primarily degenerative process, although some patients do have relapses and/or enhancing lesions. All currently approved disease-modifying therapies in MS are most active against inflammation.
Acute relapses of MS with disturbance of CNS function such as vision or mobility are thought to be periods of increased inflammatory activity of the immune system and treated accordingly.
Clinical progression, such as the gradual loss of ability to ambulate over several years, and/or poorer recovery from relapses, is believed to be a manifestation of combined ongoing chronic low-level inflammation with degenerative processes.
Brain and spinal cord magnetic resonance imaging (MRI) manifestations of inflammation show contrast-enhancing lesions with limited oedema, whereas MRI manifestations of the progressive process show atrophy and T1 hypointensity (or black holes).
Management of MS attempts to reduce the potential for triggering the bursts of inflammatory activity known as relapses, as well as limiting the extent of the relapses. Prevention or reduction of inflammation is presumed to reduce cumulative axon loss and long-term disability.
Classification
Phenotypic classification[2][3]
MS phenotypes include a consideration of disease activity (based on clinical events and imaging findings) and disease progression.
1. Relapsing disease
Clinically isolated syndrome (CIS) is a clear-cut syndrome such as optic neuritis, brainstem/cerebellar dysfunction, or partial myelitis, that does not fulfill criteria for dissemination in space and time. It is considered to be part of the relapsing MS disease spectrum. CIS may be active or not active. If a subsequent clinical event or radiological activity (gadolinium-enhancing or new/enlarging T2 lesions) follows the initial event in CIS, this becomes relapsing-remitting MS.
Relapsing-remitting MS (RRMS) requires clinical and/or magnetic resonance imaging (MRI) evidence of dissemination in space and time. RRMS is also characterised as active or not active within a specified time frame (e.g., 6 months, 1 year). As a guide, assessments for disease activity should be conducted at least annually.
2. Progressive disease
Progressive disease, whether primary progressive (progressive accumulation of disability from onset) or secondary progressive (progressive accumulation of disability after an initial relapsing course), has four possible sub-classifications taking into account the level of disability:
Active and with progression (individual has had an attack and is also gradually worsening)
Active but without progression (e.g., individual has had an attack within a previous specified time frame [i.e., 1 year, 2 years])
Not active but with progression (e.g., walking speed has decreased)
Not active and without progression (stable disease).
MS variants and related conditions
Clinically isolated syndrome (CIS) and/or monosymptomatic demyelinating event:
Monophasic demyelinating syndrome, which may or may not develop into MS, has the same demographics as RRMS.
2017 McDonald criteria state that in patients with a typical CIS and clinical or MRI demonstration of dissemination in space, the presence of cerebrospinal fluid-specific oligoclonal bands allows a diagnosis of MS; symptomatic lesions can be used to demonstrate dissemination in space or time in patients with supratentorial, infratentorial, or spinal cord syndrome; and cortical lesions can be used to demonstrate dissemination in space.[4]
Cerebrospinal fluid (CSF)-specific oligoclonal bands and the presence of T2-weighted MRI lesions at the time of the first clinical event have been identified as independent risk factors for conversion to RRMS.
Various treatment trials have indicated a delay in a second clinical event when CIS cases are treated with disease-modifying therapies used in RRMS. [
 ]
[ ]
]
[ ]
Radiologically isolated syndrome (RIS)
The term RIS is used for patients who have brain abnormalities on MRI suggestive of MS, but who have not had symptoms suggestive of a clinical event.
Neuromyelitis optica spectrum disorders (NMOSD):
NMOSD are no longer considered an MS variant because of distinctive immunopathology, MRI features, and treatment options.
Severe relapses that can be devastating, involving optic nerves and spinal cord only.
Causes loss of vision in one or both eyes, and/or long necrotic cervical and thoracic cord lesions over several segments, resulting in severe paraparesis or quadriparesis that may occur over the course of days or weeks.[5][Figure caption and citation for the preceding image starts]: Cervical spine magnetic resonance imaging scan illustrating neuromyelitis optica spectrum disorder. Extensive multiple levels of cervical spinal cord involvement with oedema and blood-brain barrier breakdown as illustrated by the contrast-enhanced T1-weighted image (left). The T2-weighted image (right) indicates the extent of signal abnormality that may manifest clinically as quadriparesis with severe spasticity and painFrom the collection of Dr Lael A. Stone [Citation ends].
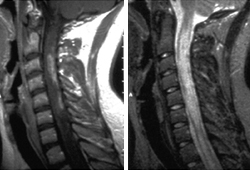
Demographics of NMOSD are different from typical MS in that there is a non-white predominance.
Treatment also differs, as this is an antibody-mediated disease amenable to immunosuppression and, in severe cases, plasma exchange.[6]
Testing for auto-antibodies associated with NMOSD (anti-aquaporin 4 ([AQP4]) and anti-myelin oligodendrocyte glycoprotein [MOG] antibodies) is required. Cell-based assays are more specific than enzyme-linked immunosorbent assay (ELISA) and should be used if available.[7][8]
Acute disseminated encephalomyelitis (ADEM):[9]
Monophasic illness that is a related, but distinct entity, to MS.
Presents with dramatic post-viral or post-vaccination dysfunction of the central nervous system (CNS) including encephalopathy and multiple brain lesions on MRI that all appear to be roughly simultaneous.
Most common in the paediatric age group, but can occur in adults.
Clinical episode may last 3 to 6 months.
No signs, symptoms, or radiographic evidence of prior or subsequent damage to the CNS.
Use of this content is subject to our disclaimer