History and exam
Key diagnostic factors
common
family history of neuropathy, pes cavus, or abnormal gait
The majority of individuals have family history of the condition; and family history of neuropathy, pes cavus (high foot arches with hammer toes), or abnormal gait strongly points to an inherited disorder, particularly Charcot-Marie-Tooth disease.
Lack of family history does not preclude the diagnosis.
walking difficulties
The most common features are weakness and atrophy of the lower leg and foot. Most people present with difficulties in walking, twisting of the ankles, and slapping of the feet.
Features are normally symmetrical, although asymmetries may occur. The majority of individuals remain ambulatory, although they may need braces and other assistive devices.
pes cavus
High-arched feet and hammer toes (pes cavus) are common but not pathognomonic, as pes cavus is seen in a variety of conditions and can be present without a neurological problem. If pes cavus is associated with areflexia, the likelihood of Charcot-Marie-Tooth disease is high. Pes cavus results from muscle imbalance as anterior tibialis and the intrinsic foot muscles are affected with sparing of the gastrocnemius muscle. The stronger pull of gastrocnemius overcomes the weaker pull of anterior tibialis, leading to structural foot deformities.[8][Figure caption and citation for the preceding image starts]: Pes cavus, toe clawing, and bilateral peroneal muscular atrophy in patient with CMT1AAdapted from Berciano J, Gallardo E, Garcia A, et al. Charcot-Marie-Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 2006 Oct;77(10):1169-76 [Citation ends].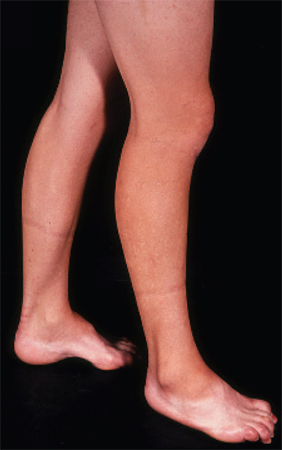
steppage gait
Assessment of the gait reveals slapping of the feet, or loss of a heel-to-toe pattern, and the person may walk with a steppage gait (lifting legs up excessively to clear the toes).
diffuse deep tendon hyporeflexia or areflexia
Deep tendon reflexes are diffusely absent (areflexia) or reduced (hyporeflexia).[8] The loss of reflexes may occur in any condition involving nerve damage, but the diffuse nature of the affected sites is not typical of other conditions. As Charcot-Marie-Tooth disease progresses in a length dependent manner, the loss of reflexes often follows the same pattern, with areflexia at the Achilles tendon being universal.
reduced muscle strength
Strength is reduced in the distal muscles of the arms and legs (e.g., intrinsic hand and foot muscles, anterior tibialis) with associated atrophy and weakened foot eversion. This clinical observation points to a length-dependent lower motor neuron disorder.
The most distal nerves degenerate (axonal loss) first. Thus, the most distal muscles are affected first, so that the feet are affected before the ankles, which are in turn affected before the hands, and strength is typically maintained in the proximal muscles and the gastrocnemius muscle strength is usually greater in comparison to the anterior tibialis.[8][Figure caption and citation for the preceding image starts]: Wasting of hand muscles in patient with CMT1AAdapted from Berciano J, Gallardo E, Garcia A, et al. Charcot-Marie-Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 2006 Oct;77(10):1169-76 [Citation ends].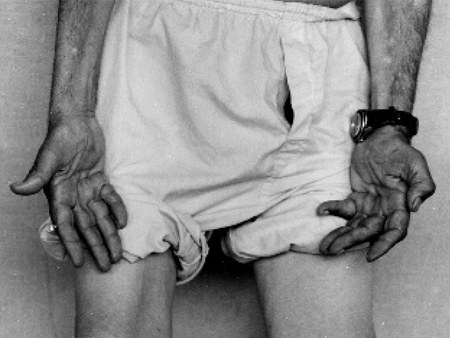 [Figure caption and citation for the preceding image starts]: Pes cavus, toe clawing, and bilateral peroneal muscular atrophy in patient with CMT1AAdapted from Berciano J, Gallardo E, Garcia A, et al. Charcot-Marie-Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 2006 Oct;77(10):1169-76 [Citation ends].
[Figure caption and citation for the preceding image starts]: Pes cavus, toe clawing, and bilateral peroneal muscular atrophy in patient with CMT1AAdapted from Berciano J, Gallardo E, Garcia A, et al. Charcot-Marie-Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 2006 Oct;77(10):1169-76 [Citation ends].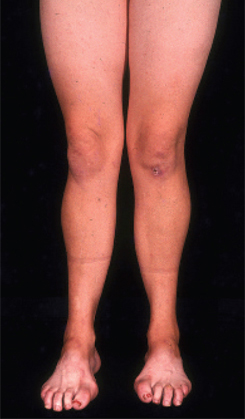
uncommon
transient sensory symptoms
Hereditary neuropathy with liability to pressure palsies presents with transient sensory and motor symptoms after minor compression or stretching of a nerve. These symptoms can last for weeks to months and are usually asymmetrical.
transient motor symptoms
Hereditary neuropathy with liability to pressure palsies presents with transient sensory and motor symptoms after minor compression or stretching of a nerve. These symptoms can last for weeks to months and are usually asymmetrical.
Other diagnostic factors
common
past surgery to feet and ankles
Tendon transfers in the feet are common, owing to tight heel cords. Other surgeries include hammer toe straightening and triple arthrodesis of the ankle.
balance difficulties in childhood
There may be a history of difficulty with balance or skating as a child, and problems in finding well-fitting shoes owing to high foot arches.
ankle weakness
Nerves to the anterior tibialis muscle are preferentially affected, causing ankle weakness and loss of the ability to dorsiflex the foot.[8]
sensory abnormalities in hands and feet
The most distal nerves are affected first, and abnormal sensations, such as loss of sensation and burning or tingling in the hands and feet, typically begin at the toes and proceed proximally over time.
toe-walking
Toe-walking and the absence of heel-walking result from a tight heel cord, due to the nerves to anterior tibialis being preferentially affected, leading to inadequate strength to pull the foot up during ambulation.[8]
uncommon
delayed motor milestones
Children with delayed motor milestones, who never ran or ambulated, have a severe early-onset phenotype of CMT1, CMT2, and CMT4 called Dejerine-Sottas syndrome. Late-normal age of walking is common (12-14 months).
sensory ataxia
Imbalance and in-coordination due to loss of proprioception may be present.
kyphoscoliosis
Curvature of the spine (kyphoscoliosis) can occur in some individuals with Charcot-Marie-Tooth disease and is a prominent feature in some forms (e.g., CMT4C due to mutations in SH3TC2).
Risk factors
strong
family history of neuropathy, pes cavus (high foot arches with hammer toes), or abnormal gait
Charcot-Marie-Tooth (CMT) disease is a genetic condition and there are no known risk factors for the condition, other than a family history. Depending on the inheritance pattern of the CMT subtype, the risk of passing the condition on to the next generation differs.
In autosomal dominant forms (most CMT1 and CMT2), one parent with the genetic disorder is sufficient to pass on the condition, and each child has a 50% chance of inheriting the disorder. In autosomal recessive forms (CMT4), both parents carry one genetic mutation, but do not have clinical manifestations of the disease, as they have one working copy of the gene. In such cases, each child has a 25% chance of getting the disorder, a 50% chance of being a carrier, and a 25% chance of inheriting two working copies of the gene and being an unaffected non-carrier.
Females with an X-linked form of CMT have a 50% chance of passing on the condition, and males with an X-linked form have a 100% chance of passing on the condition to daughters but a 0% chance of passing it on to sons.
Use of this content is subject to our disclaimer