Approach
There are several features characteristic of Charcot-Marie-Tooth (CMT) disease.[8] Affected individuals have typically had difficulty since childhood with balance and with weakness in the ankles. Deep tendon reflexes are usually absent or decreased diffusely in both upper and lower extremities, although the most consistent site is at the Achilles tendon. Nerve conduction studies will show either a reduction in velocity, with decreased amplitudes and prolonged distal latencies in demyelination in CMT1, or decreased amplitudes with normal velocities in the presence of axonal loss in CMT2.
The presence of a peripheral neuropathy with a positive family history and/or abnormal genetic testing is diagnostic. When there is no family history of CMT and genetic testing for the condition is negative, it is often difficult to distinguish between CMT and an acquired inflammatory neuropathy. In these situations, sural nerve biopsy can be used to detect the presence of other disorders instead of CMT.
The severity of CMT can be determined using the Charcot-Marie-Tooth Neuropathy Score (CMTNS), a validated measure of impairment for people with the condition.[9] The CMTNS was updated in 2011, creating the CMTNSv2, which decreased ceiling effects of the CMTNS.[10] On a 36-point scale that includes symptoms, clinical findings, and electrophysiology, 0-10 points represents mild impairment, 11-20 points moderate impairment, and ≥21 points severe impairment. A subset of this scale, called the CMT Examination Score (CMTES), was updated in 2020 using Rasch analysis, and can provide a quick analysis of how an individual is doing clinically, using the signs and symptoms of the CMTNS, but not the electrophysiology scores.[11]
Clinical history
To determine if a person has CMT, it is important to obtain a thorough clinical history, including detailed past medical, developmental, and family histories.
Presenting features
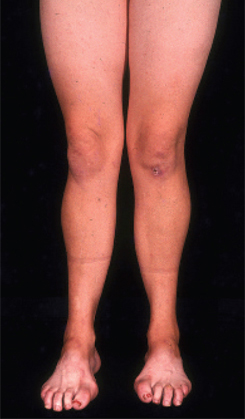
The most common features are weakness and atrophy of the lower leg and foot.[Figure caption and citation for the preceding image starts]: Pes cavus, toe clawing, and bilateral peroneal muscular atrophy in patient with CMT1AAdapted from Berciano J, Gallardo E, Garcia A, et al. Charcot-Marie-Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 2006 Oct;77(10):1169-76 [Citation ends].
 Most individuals present with difficulties in walking, twisting of the ankles, and slapping of the feet. Nerves to the anterior tibialis muscle are preferentially affected, causing ankle weakness and loss of the ability to dorsiflex the foot.[8] Features are normally symmetrical, although asymmetries may occasionally occur.
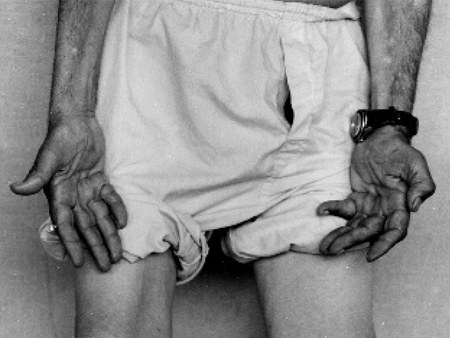
Most individuals present with difficulties in walking, twisting of the ankles, and slapping of the feet. Nerves to the anterior tibialis muscle are preferentially affected, causing ankle weakness and loss of the ability to dorsiflex the foot.[8] Features are normally symmetrical, although asymmetries may occasionally occur.Symptoms occur in a length-dependent manner, as the most distal nerves degenerate (axonal loss) first. Thus, the most distal muscles are affected first, so that the feet are affected before the ankles, which are in turn affected before the hands.[8] Abnormal sensations, such as loss of sensation and burning or tingling in the hands and feet, typically begin at the toes and proceed proximally over time.[Figure caption and citation for the preceding image starts]: Wasting of hand muscles in patient with CMT1AAdapted from Berciano J, Gallardo E, Garcia A, et al. Charcot-Marie-Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 2006 Oct;77(10):1169-76 [Citation ends].
 [Figure caption and citation for the preceding image starts]: Pes cavus, toe clawing, and bilateral peroneal muscular atrophy in patient with CMT1AAdapted from Berciano J, Gallardo E, Garcia A, et al. Charcot-Marie-Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 2006 Oct;77(10):1169-76 [Citation ends].
[Figure caption and citation for the preceding image starts]: Pes cavus, toe clawing, and bilateral peroneal muscular atrophy in patient with CMT1AAdapted from Berciano J, Gallardo E, Garcia A, et al. Charcot-Marie-Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 2006 Oct;77(10):1169-76 [Citation ends].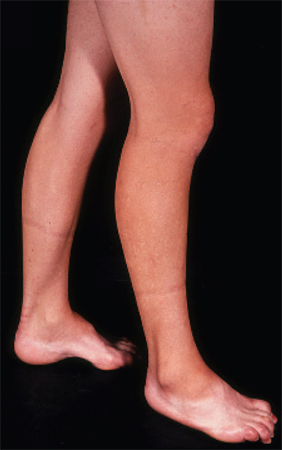
Children with delayed motor milestones, who never ran or ambulated, have a severe early-onset phenotype of CMT1, CMT2, and CMT4 called Dejerine-Sottas syndrome. Hereditary neuropathy with liability to pressure palsies (HNPP) presents with transient sensory and motor symptoms after minor compression or stretching of a nerve. These symptoms can last for weeks to months and are usually asymmetrical. Most people with CMT2A have significant motor weakness in the pre-school and school-age years, and will be non-ambulatory by the age of 20 years, although sensory impairment is not affected until later in the disease course. Proximal muscle weakness may also occur in these individuals. Curvature of the spine (kyphoscoliosis) can occur in some people with CMT and is a prominent feature in some forms (e.g., CMT4C due to mutations in SH3TC2).
Past medical history
Key questions in the developmental history include difficulty with balance or skating as a child, and problems in finding well-fitting shoes owing to high foot arches.
Past surgical procedures to the feet and ankles should also be noted. Tendon transfers in the feet and Achilles tendon lengthening are common surgeries performed for tight heel cords. Other surgeries include hammer toe straightening, and triple arthrodesis of the ankle.
Family history
As CMT is a hereditary peripheral neuropathy, it is important to obtain a family history going back at least three generations. The majority of affected individuals have a family history of the condition; and a family history of neuropathy, pes cavus (high foot arches with hammer toes), or abnormal gait strongly points to an inherited disorder, particularly CMT.
It is important to draw a family tree (pedigree) and track both sides of the family for three generations to determine who is affected and what mode of inheritance is present. Questions such as, "did/does this relative have any trouble with walking, balance, tripping, falling, or with their hands or sensations?" should be asked. An autosomal dominant pedigree will have people affected in every generation, and male-to-male transmission can be present. X-linked pedigrees will not have male-to-male transmission, and males are almost always more severely affected than females. Autosomal recessive pedigrees may have only one person or sibling affected.
Lack of a family history does not preclude the diagnosis, as de novo mutations are relatively common.[12][13] There is also phenotypic variability such that parents or other family members may be unaware of their disease, and recessive cases can present with no family history.
Physical examination
The neurological examination should test the cranial nerves, strength, sensation, reflexes, co-ordination, and ambulation.
The cranial nerve examination is typically normal. Some individuals with CMT1B have abnormalities of pupillary constriction, and people with CMT2A may have optic nerve abnormalities.
Sensory ataxia (i.e., imbalance and in-coordination due to loss of proprioception) may be present.
Strength is reduced in the distal muscles of the arms and legs (e.g., intrinsic hand and foot muscles, anterior tibialis), with associated atrophy and weakened foot eversion. This clinical observation points to a length-dependent lower motor neuron disorder. Strength is typically maintained in the proximal muscles and in the gastrocnemius muscle.[8][Figure caption and citation for the preceding image starts]: Wasting of hand muscles in patient with CMT1AAdapted from Berciano J, Gallardo E, Garcia A, et al. Charcot-Marie-Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 2006 Oct;77(10):1169-76 [Citation ends].
[Figure caption and citation for the preceding image starts]: Pes cavus, toe clawing, and bilateral peroneal muscular atrophy in patient with CMT1AAdapted from Berciano J, Gallardo E, Garcia A, et al. Charcot-Marie-Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 2006 Oct;77(10):1169-76 [Citation ends].Sensation corresponding to both small and large sensory nerve fibres is decreased or absent.[8] There is a reduction in pinprick and vibration sensation, which is more pronounced at the toes than in more proximal regions.[8]
Deep tendon reflexes are diffusely absent (areflexia) or reduced (hyporeflexia).[8] The loss of reflexes may occur in any condition involving nerve damage, but the diffuse nature of the affected sites is not typical of other conditions.
High-arched feet and hammer toes (pes cavus) are common but not pathognomonic, as pes cavus is seen in a variety of conditions and can be present without a neurological problem. If pes cavus is associated with areflexia, the likelihood of CMT is high. Pes cavus results from muscle imbalance as anterior tibialis and the intrinsic foot muscles are affected with sparing of the gastrocnemius muscle. The stronger pull of gastrocnemius overcomes the weaker pull of anterior tibialis, leading to structural foot deformities.[8][Figure caption and citation for the preceding image starts]: Pes cavus, toe clawing, and bilateral peroneal muscular atrophy in patient with CMT1AAdapted from Berciano J, Gallardo E, Garcia A, et al. Charcot-Marie-Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 2006 Oct;77(10):1169-76 [Citation ends].
Assessment of the gait reveals slapping of the feet, or loss of a heel-to-toe pattern, and the affected person may walk with a steppage gait (lifting legs up excessively to clear the toes). Toe-walking and the absence of heel-walking result from a tight heel cord, which is due to the nerves to anterior tibialis being preferentially affected, leading to inadequate strength to pull the foot up during ambulation.[8] Patients may require bilateral aids, such as ankle-foot orthoses, to ambulate.
Imaging
A hip x-ray should be considered in all children to screen for hip dysplasia.[14] X-rays of cervical, thoracic, and lumbar spine and pelvis (spinal series) should be considered for children exhibiting signs of scoliosis or kyphosis. Postero-anterior and lateral views are recommended.
Pulmonary function testing
Baseline pulmonary function testing should be undertaken in the following children when they are able to complete this reliably (usually from the age of 5-6 years):[14]
Children with symptoms of sleep-disordered breathing (e.g., unexplained headaches, daytime somnolence or symptoms of obstructive sleep apnoea).
Children with recurrent lower respiratory tract infections (>2 courses of antibiotics over 4 months or >2 hospital admissions for a respiratory tract infection in 12 months).
Children with scoliosis (Cobb angle of >40°).
All non-ambulant children.
Nerve conduction studies (NCS)
This investigation should be considered for any individual with a suspected neuropathy, as it can confirm the diagnosis of a polyneuropathy and determine if the disorder is primarily motor, sensory, or mixed, and if it affects myelin and the Schwann cell, or the axon. Both motor and sensory nerves should be studied, although it should be noted that interpretation of the results can be difficult, as some forms of CMT show only slight abnormalities on NCS.
The nerve abnormalities are symmetrical, with a similar degree of slowing or reduction in amplitude in all the tested nerves, and sensory nerve responses are typically absent. Demyelination in CMT1 (including CMT1A) is associated with very slow conduction velocities of 15-38 m/second, reduced or absent sensory responses, and prolonged distal latencies. Axonal loss in CMT2 (including CMT2A) is associated with reduced amplitudes (particularly in CMT2A), but with relatively normal conduction velocities of >38 m/second.
In the presence of extreme atrophy in the distal muscles, responses may not be recordable, and it may be necessary to perform the NCS at a more proximal muscle than would typically be used.
Genetic testing
There are more than 90 known genetic causes, and commercial testing is available in the US for most of these.
Genetic testing should be done only after appropriate counselling. The rationales of determining the specific genetic aetiology of the disease include psychological comfort, family planning, the ability to participate in clinical trials, and providing a better knowledge of the natural history of the particular CMT subtype. These considerations should be weighed against potential for discrimination in jobs and insurance, the cost to the individual, the stigma associated with a genetic diagnosis, and family dynamics.
A logical approach based on nerve conduction testing and a genetic history can streamline the genetic testing.[7][15] Although over 90 genes are known to cause CMT when mutated, about 92% of people with an identifiable mutation will have a mutation in 1 of 4 genes: PMP22 (duplication or deletion), GJB1, MPZ, or MFN2.[16] Phenotypes can help narrow down the subtype of CMT, using the part of the nerve affected (myelin or axon), the mode of inheritance (autosomal dominant, autosomal recessive, or X-linked), and the age of onset (infancy, childhood, adult).[7] Furthermore, genetic testing can be streamlined based on prevalence numbers. Persons with a family pedigree showing a dominant mutation or male-to-male transmission and signs of demyelination on NCS should be tested for CMT1A, the most common form of demyelinating CMT. If there is no duplication of the PMP22 gene indicative of CMT1A, testing for other myelin gene disorders may be appropriate. If an X-linked disorder is possible, testing for CMT1X with mutations of the GJB1 gene is appropriate. A causative gene mutation is identified in approximately 80% of cases of demyelinating CMT.[17] Persons with an axonal pattern on NCS, with or without a family history, should be tested for mutations in MFN2 causing CMT2A, the most common form of axonal CMT.[13] Only 30% to 40% of cases of axonal CMT will have an identifiable mutation.
Next Generation Sequencing panels allow for rapid and cheap options for testing multiple genes at one time. Nerve conduction studies, family history, and physical examination can help with interpretation of large panel results. For example, if a variant is found in an axonal gene, but the individual has demyelinating CMT, it is unlikely that the axonal gene variant is disease-causing. Exome sequencing is quickly becoming less expensive and more useful in finding new genes in CMT. If a person has negative genetic testing for the most likely known causes of CMT, it may be reasonable to send their DNA and DNA of other affected family members for research or clinical exome testing.
Result reports should be interpreted with caution. The detection of a known gene mutation associated with a dominantly inherited form of CMT is definitely the cause of the condition. However, a single disease-causing mutation in a recessive gene cannot be the cause. Genetic variants of uncertain significance are typically amino acid-changing mutations that have not yet been classified as disease-causing or benign, and can usually only be interpreted by testing the parents and tracking to see if the condition and the variant segregate together. It is always appropriate to call the laboratory that performed the test to ask for clarification and interpretation of the results.
It is recommended to consult with a specialist in a centre that sees many people with CMT to arrange the most efficient and accurate testing. Many specialists would first do a panel and then proceed to exome sequencing if needed.
Use of this content is subject to our disclaimer