Approach
RP is a lifelong incurable disease that leads to progressive loss of vision. Due to the implications of such a diagnosis, it is important that patients with suspected RP be seen by a consultant. If RP is confirmed then syndromic associations should be considered.
History
Visual loss is gradual and progressive. Visual acuity varies from perfect 20/20 vision to merely light detection. It is unusual, however, to have total visual loss, and some degree of sight is usually maintained into old age. Central acuity can be decreased in advanced forms of the more common rod-cone dystrophy form of the disease or at an earlier time if cystoid macular oedema (CMO) develops.
Symptoms depend on which types of photoreceptors are affected. Most commonly, the rods are affected first, and some of the earliest symptoms include difficulty driving at night due to poor vision and impaired adaptation when entering a dark room. Loss of peripheral vision is another feature, and the patient may report bumping into furniture or the edge of doors or having difficulty playing racket sports. Visual field loss often goes unnoticed until it reaches a moderate stage due to the overlapping visual fields of each eye.
In cone-rod-type RP, where the cones are predominantly affected first, symptoms may include difficulty in reading or seeing detail (due to central visual loss) or impaired colour vision. Patients may report seeing flashes of light or luminous rays, although these may also be seen with migrainous aura or retinal detachment. However, with RP, the flashes are continual rather than episodic. Glare from bright lights may be a problem in advanced disease.
A severe subtype of RP, known as Leber's congenital amaurosis, can present in infancy with decreased vision, sluggish pupils, and nystagmus.[5]
Many patients with X-linked or dominant RP have a family history of the disease. Autosomal recessive inheritance may or may not reveal a family history. As X-linked forms only affect male offspring (although females can be carriers), this may not be obvious if there were no male children within a generation. X-linked forms of the disease usually present in childhood, while autosomal recessive and dominant forms tend to present later in life.[4]
Syndromic RP
RP is most often found in isolation but also can exist as part of a syndrome. To exclude the presence of syndromic disease, it is important to look for other symptoms and signs: in particular, hearing or balance problems (Usher's, Alstrom's), obesity (Bardet-Biedl), renal failure (Bardet-Biedl, Alstrom's, Senior-Loken), extra digits on the hands or toes (Bardet-Biedl, Joubert's), ataxia (Joubert's, Bardet-Biedl), seizures (neuronal ceroid-lipofuscinosis), and diabetes (Alstrom's disease). Hearing loss, ataxia, ophthalmoplegia, cardiac conduction defects, and dysphagia may be seen in Kearns-Sayre syndrome. Abetalipoproteinuria is a potentially reversible cause of RP, as many of the features are due to lack of absorption of fat-soluble vitamins. If diagnosed early, retinal problems can be prevented or slowed by supplementation with vitamins A and E. Infantile Refsum's disease usually presents in early childhood with accompanying hearing impairment, co-ordination problems, and poor muscle tone. In adult Refsum disease, features may include anosmia, ataxia, and cardiac arrhythmias. Referral to a geneticist may be required for syndromic diagnosis.
Eye examination
The American Academy of Ophthalmology Preferred Practice Pattern Committee reported on recommendations for the frequency for adult comprehensive medical eye examinations for asymptomatic patients, patients with, or without risk factors for eye disease.[22]
Visual acuity
This may be measured using a Snellen chart and can be varied depending on the type and severity of RP. It is essential to record visual acuity for both functional and legal implications and to track progress. Refractive errors may be noted and sight may be improved by glasses if this is corrected, although underlying RP remains. In early severe forms of RP, such as Leber's congenital amaurosis, hyperopia predominates. In later-onset forms of RP, myopia and astigmatism are common. About half of patients with RP will develop cataracts, and removal may improve vision if underlying RP has not progressed too far.
Funduscopy
Typically the optic disc is pale and the optic nerve has a waxy appearance, although it may appear normal in early disease.
Vascular attenuation of retinal vessels helps to distinguish RP from choroideraemia (which demonstrates normal retinal vessels but atrophy of the choroidal vessels). [Figure caption and citation for the preceding image starts]: Waxy pallor and vascular attenuationFrom the Oregon Retinal Degeneration Center collection [Citation ends].
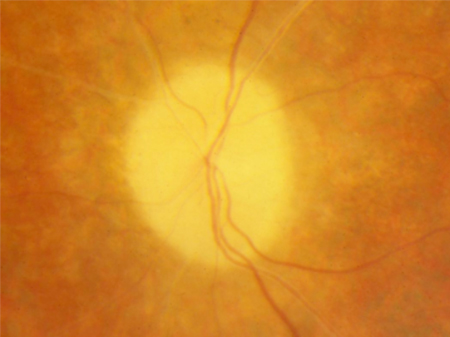
Retinal pigment epithelium degeneration is a key feature. Bone spicule pigmentation results from migration of retinal pigment epithelial cells into the retina, often surrounding retinal vessels. The appearance is of black dots or clumps of dots on the retina.[Figure caption and citation for the preceding image starts]: Bone spiculesFrom the Oregon Retinal Degeneration Center collection [Citation ends].
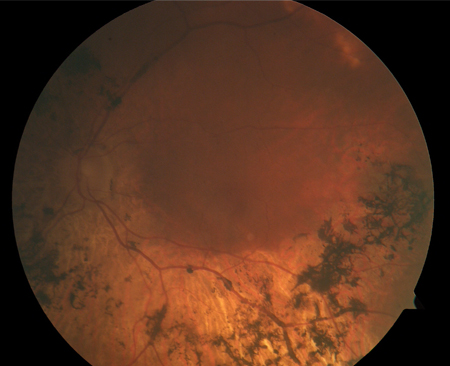 [Figure caption and citation for the preceding image starts]: Retinitis pigmentosaFrom the Oregon Retinal Degeneration Center collection [Citation ends].
[Figure caption and citation for the preceding image starts]: Retinitis pigmentosaFrom the Oregon Retinal Degeneration Center collection [Citation ends].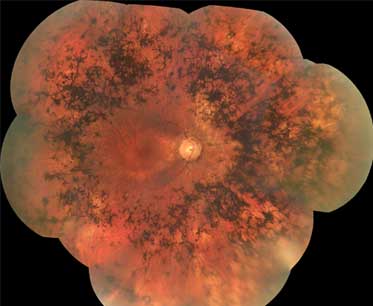
Optic nerve head drusen: optic disc drusen are congenital and developmental anomalies of the optic nerve head. They are formed by calcific degeneration in some of the axons of the optic nerve. They are often an incidental finding but are more common in patients with RP. With time they can grow and impinge on the retinal nerve fibre layer and cause visual field defects. A small cup-to-disc ratio may indicate buried optic drusen.
CMO: this is a common complication of RP and it appears like a tiny cyst in the fovea when viewed on high magnification of the fundus using a slit lamp. These are unlikely to be visualised by the non-ophthalmologist.
Mild vitreous cells: these may be observed when viewed by an expert on slit-lamp examination. More significant inflammation should prompt suspicion for other diseases that can mimic RP.
Coats-like retinopathy: a rare sign seen in certain patients with RP. It is characterised by peripheral areas of retina with abnormal vascular telangiectasia, which can leak and cause retinal exudation.
Keratoconus and glaucoma
More common in patients with RP than in the general population.
Keratoconus is visual distortion due to structural changes in the cornea, which gives it a more conical appearance. This can affect visual acuity, and night vision is often particularly poor.
Glaucoma is the result of either acute or chronic elevation of intraocular pressure causing damage to the optic nerve. It can be detected by measuring intraocular pressure.
Investigations
The number of tests varies between centres, but all patients with suspected RP should have visual fields tested and an electroretinogram (ERG).
Examination of visual fields: can be performed in different ways, and tests depend on whether the stimulus is static or moving (kinetic). The Goldmann kinetic perimetry test has historically been the method for assessing a patient's full visual field. It provides essential information regarding function, such as ability to drive safely. However, Octopus 900 perimetry is now capable of performing both kinetic and static full field perimetry. Mid-peripheral visual field defects are one of the pathognomonic features of RP. Defects can start as islands in the mid-periphery, expand to form crescents, and finally result in a complete ring scotoma. In time, these ring scotomas can enlarge, ultimately leaving patients with a diminishing tunnel field of vision.
Full field ERG: this measures the electrical response of cells in the retina, including photoreceptors. It involves placing electrodes on the cornea or skin around the eye and measuring the amplitude of responses to standard stimuli such as a flash of light. Abnormal ERGs are an essential feature of RP. A decrease in amplitude and an increase in latency can be seen in both the dark-adapted and light-adapted ERGs.
Further tests that may be carried out to confirm the diagnosis include elevated dark-adapted threshold and optical coherence tomography (OCT). These are not carried out routinely at all centres.
Elevated final dark-adapted threshold involves placing a patient in a completely dark room and determining the dimmest light that can be perceived. It replicates symptomatic reports of difficulty adapting to darkened environments.
OCT of the retina is a non-invasive imaging system using the detection of optical light reflection to build up a 3-dimensional picture. It is non-contact and does not involve any radiation exposure. It can reveal retinal atrophy and is the preferred method for determining the presence of CMO. This should be considered in any patient with decreased central visual acuity suggestive of CMO.
Adaptive optics imaging is new technology that allows for high-resolution imaging of the photo-receptor mosaic by compensating for corneal and lenticular aberrations during imaging. Custom-built systems are capable of resolving individual cones and rods in some individuals.
Genetic testing for RP involves drawing blood from the patient and sending it to a laboratory that tests for specific mutations for this disease. Genetic testing is available for only a subset of the total genes in RP, and even testing of known genes is not always completely sensitive. It is most effective when a particular gene is suspected. This is only performed in certain people after consultation with a clinical geneticist when the probability of identifying the responsible gene is good. This would confirm the diagnosis. Newer technologies using whole exome sequencing are now becoming available and allow testing of many genes at one time. Additionally, these technologies offer the hope of finding new genes for RP.
Wide-field fundus autofluorescence (FAF), an imaging modality, identifies areas of the fundus with irregular distribution of lipofuscin and other fluorophores in the retinal pigment epithelium (RPE) cell monolayer. Patterns of irregular FAF in the posterior pole and peripheral retina are associated with specific hereditary retinal degenerations, and may be useful in monitoring disease progression and response to novel therapies. Guidelines from the American Academy of Ophthalmology give recommendations for the clinical assessment of patients with inherited retinal degenerations.[23]
Use of this content is subject to our disclaimer