Monitoring
Regular screening should be carried out, including:[5]
Thyroid function tests (TFTs) for autoimmune thyroid disease annually; antithyroid antibodies may be measured if TFTs are abnormal
LFTs to screen for 'Turner hepatitis' after the age of 10 years and repeated annually
LH and FSH to be repeated annually
BP measurements for hypertension at every consultation
Fasting blood sugar and HbA1c for diabetes risk repeated annually after the age of 10 years
Blood lipids to monitor dyslipidaemia repeated annually after the age of 18 years if there is at least one risk factor for cardiovascular disease
Screening for conductive and/or sensorineural hearing loss, to be repeated every 3 years in childhood and every 5 years in adulthood[5]
Screening for refractive errors and visual abnormalities[5]
Strabismus, amblyopia, hyperopia and myopia, ptosis and colour blindness have all been reported. Girls with Turner Syndrome have an epicanthal fold (upper eyelid fold that covers inner corner of eye) and hypertelorism (increased distance between eyes). A comprehensive ophthalmological examination should be performed between aged 12 and 18 months or at the time of diagnosis, if at an older age.
Screening for congenital renal anomalies[5]
At the time of diagnosis, a renal ultrasound is done to assess the presence of structural abnormalities, such as horseshoe kidney, renal agenesis, and duplicated collecting system, which affects approximately 25% of patients with Turner syndrome.[3][4]
Renal function is usually normal, but obstruction of the collecting system is associated with urinary infection and may need correction.
Screening for congenital cardiovascular defects
Aortic coarctation and bicuspid aortic valves are the most common abnormalities. Other cardiac abnormalities include partial anomalous pulmonary veins, left heart hypoplasia, and a dilated aorta. Prevalence of cardiovascular defects is much higher in those with clear evidence of fetal lymphoedema, such as neck webbing.
A functionally bicuspid valve as a result of complete or partial fusion of the right and left coronary leaflets is seen in 30% of asymptomatic patients, and poses a risk for infection, valve deterioration, and aortic dilation and dissection.[18] Congenital cardiovascular defects constitute the major source of premature mortality in Turner syndrome. Two genes,TIMP1 and TIMP3, which are both located on the short arm of the X chromosome when hemizygous increase the risk of aortopathy with an odds ratio of 12.86.[19]
Visualisation of the aortic valve, thoracic aorta, and pulmonary veins should be attempted by transthoracic echocardiography (TTE) in infants or by cardiac magnetic resonance (CMR) imaging or CT in older children and adults. If no anomalies are detected and systemic BP is normal, re-evaluation of the cardiovascular system with either TTE or CMR should be performed again every 5-10 years during adulthood, especially prior to desiring pregnancy or with the development of hypertension.
Baseline bone mineral density should be assessed at the transition to adult care and re-evaluated every 5 years in adults and when discontinuing oestrogen.[5]
Tissue transglutaminase IgA level (if normal IgA levels) should be measured every 2 years from the age of 2, and in adults when there are suggestive symptoms, to monitor for coeliac disease.[5]
Cardiac evaluation should be carried out with TTE or CMR every 1-5 years depending on severity or as indicated.[5]
Congenital cardiovascular defects constitute the major source of premature mortality in Turner syndrome. All girls by the age of 12 years should have a screening cardiac MRI, thorough characterisation of aortic valve structure and aortic diameters, and investigation of other common anomalies. Even if no anomalies are detected and systemic BP is normal, re-evaluation of the cardiovascular system should be performed every 10 years again, especially prior to desiring pregnancy or with the development of HTN. If congenital defects are found, follow-up and treatment are dictated by the individual defect. Children should be transitioned to an adult congenital heart disease clinic, because they are at continued risk of aortic complications during adult years. The following algorithms, from the American Heart Association’s guidelines for cardiovascular health in TS, will aid in the investigation and follow-up.[5][Figure caption and citation for the preceding image starts]: Algorithm for screening and monitoring congenital cardiovascular disease in Turner syndrome in girls aged <15 yearsAdapted from: Gravholt CH, Andersen NH, Conway GS, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017;177(3):G1-G70. [Citation ends]. [Figure caption and citation for the preceding image starts]: Algorithm for screening and monitoring congenital cardiovascular disease in Turner syndrome in women and girls aged >15 years. (CHD, congenital heart disease; AV, aortic valve; BAV, bicuspid aortic valve; PAPV, partial anomalous pulmonary veins; Coarc, coarctation; ACH, adult congenital heart; CVS, cardiovascular system; HTN, hypertension)Adapted from: Gravholt CH, Andersen NH, Conway GS, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017;177(3):G1-G70. [Citation ends].
[Figure caption and citation for the preceding image starts]: Algorithm for screening and monitoring congenital cardiovascular disease in Turner syndrome in women and girls aged >15 years. (CHD, congenital heart disease; AV, aortic valve; BAV, bicuspid aortic valve; PAPV, partial anomalous pulmonary veins; Coarc, coarctation; ACH, adult congenital heart; CVS, cardiovascular system; HTN, hypertension)Adapted from: Gravholt CH, Andersen NH, Conway GS, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017;177(3):G1-G70. [Citation ends].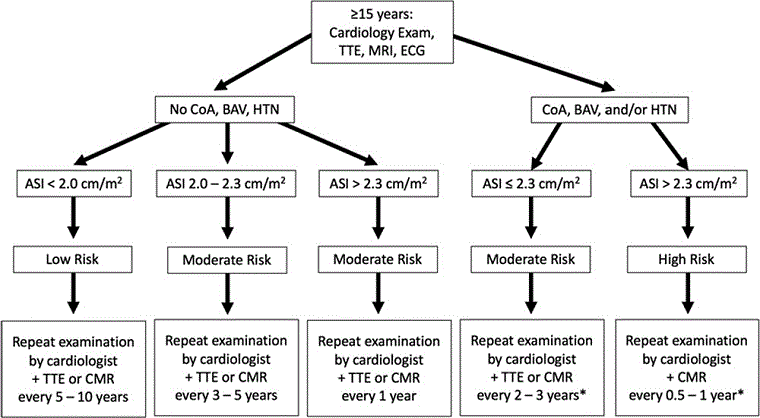
Use of this content is subject to our disclaimer