Aetiology
Genetics
A mutation in both alleles of the RB1 gene is widely believed to be a prerequisite for the presence of disease. Other mutations are likely to be necessary for progression to clinical retinoblastoma.[24]
One study showed that gene expression profiles may be dynamic, varying temporally and regionally (apical and basal tumour regions).[25] However, only 10% of patients have a previously established family history of the disease.[24][26]
The majority of cases occur as a result of spontaneous mutations either early in embryogenesis or de novo in one set of parental germline cells.[26]
Viral exposure
Human papillomavirus (HPV) types 16 and 18 have been reported in retinoblastoma samples.[27][28] Further research is required to determine whether there is a significant association between HPV and retinoblastoma.[29][30]
Advanced paternal age
Pathophysiology
Whether inherited in the germline or occurring spontaneously, retinoblastoma develops from a critical mutation in the RB1 gene, which is primarily expressed in horizontal interneurons located within the inner nuclear layer of the retina.[36] This mutation leads to the loss of heterozygosity and subsequent inactivation of RB1 (i.e., the formation of a non-functional homozygous null mutant).[24]
The RB1 gene plays a pivotal role as a tumour suppressor, regulating the cell cycle by inhibiting transition from the G1 phase to the S phase.[37][38] Its loss removes a key checkpoint in cellular replication, resulting in unchecked cellular proliferation. The presence of other members of the RB protein family, such as p107 and p130, and unrelated proteins like senescence-associated proteins, can partially compensate for the loss of RB1 and lead to delayed tumour progression in some cases.[39] However, these compensatory mechanisms are often insufficient to prevent tumourigenesis.
The lack of functional RB1 creates a cascade of downstream effects, including genomic instability, aberrant chromosomal segregation, and disruptions in the expression of proteins critical for cell cycle control, such as E2F transcription factors.[37][39] This destabilisation fosters an environment conducive to oncogenesis. As mutations accumulate, retinoblastoma cells acquire additional characteristics of malignancy, such as resistance to apoptosis, angiogenesis, and evasion of immune surveillance.
Over time, these cellular changes culminate in the clinical manifestation of retinoblastoma, which can be detected through various diagnostic techniques, including fundus examination, imaging, and genetic testing.[40]
[Figure caption and citation for the preceding image starts]: Histopathology of retinoblastoma. This image demonstrates the classical features of retinoblastoma, including densely packed, small, round tumor cells with hyperchromatic nuclei and scant cytoplasm, arranged in sheets. The absence of Flexner-Wintersteiner rosettes in this specimen does not preclude the diagnosis, as their presence is not obligatory. These histopathological features are typical of this aggressive retinal tumor and provide critical information for diagnosis, staging, and prognosisPR J. L. Kemeny, ISM/Science Photo Library; used with permission [Citation ends]. [Figure caption and citation for the preceding image starts]: Light micrograph of a retinal section from a patient with retinoblastoma, a rare form of intraocular cancer. The tumor shows disrupted retinal architecture and infiltrative growth of atypical cells. Immunohistochemistry with anti-rhodopsin antibodies highlights areas of preserved photoreceptor differentiation within the retinal tissue. This staining aids in identifying residual retinal layers amidst the malignant cellular proliferationPR J. L. Kemeny, ISM/Science Photo Library; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Light micrograph of a retinal section from a patient with retinoblastoma, a rare form of intraocular cancer. The tumor shows disrupted retinal architecture and infiltrative growth of atypical cells. Immunohistochemistry with anti-rhodopsin antibodies highlights areas of preserved photoreceptor differentiation within the retinal tissue. This staining aids in identifying residual retinal layers amidst the malignant cellular proliferationPR J. L. Kemeny, ISM/Science Photo Library; used with permission [Citation ends].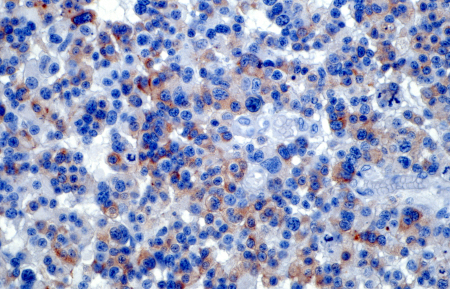
Classification
International Classification of Retinoblastoma[6]
The goal of this classification is to reflect the likelihood of ocular survival based on modern treatment techniques (typically chemotherapy plus focal therapy).
Group A
Retinoblastoma 3 mm or less in basal dimension or thickness, located at least 3 mm from the foveola and 1.5 mm from the optic nerve.
Group B
Retinoblastoma not in Group A with one or more of the following:
Macular location (≤3 mm to foveola)
Juxta-papillary location (≤1.5 mm to optic nerve)
Additional subretinal fluid (≤5 mm from margin).
Group C
Retinoblastoma tumour with one of the following:
Focal subretinal seeds
Focal vitreous seeds
Both focal subretinal and vitreous seeds.
Group D
Retinoblastoma tumour with one of the following:
Diffuse subretinal seeds
Diffuse vitreous seeds
Both diffuse subretinal and vitreous seeds.
Group E
Very high-risk eyes with one or more of the following:
Neovascular glaucoma
Massive intraocular haemorrhage
Aseptic orbital cellulitis
Tumour anterior to the vitreous face
Tumour touching the lens
Diffuse infiltrating retinoblastoma
Phthisis bulbi (also known as end-stage eye, this is a non-functioning, atrophic, scarred, and disorganised globe, frequently with dystrophic calcification).
Reese-Ellsworth's classification[7]
Reflects the likelihood of ocular survival after lateral port external beam radiation. The Reese-Ellsworth classification has largely been superseded by the International Intraocular Retinoblastoma Classification (following the introduction of intravenous chemotherapy for intraocular retinoblastoma).
Group I
a. Solitary tumour, <4 disc diameters in size, at or posterior to the equator of the eye (an imaginary line in the coronal plane that marks the division between the anterior and posterior halves of the eye).
b. Multiple tumours, none >4 disc diameters in size, all at or behind the equator.
Group II
a. Solitary tumour, 4 to 10 disc diameters in size, at or behind the equator.
b. Multiple tumours, 4 to 10 disc diameters in size, behind the equator.
Group III
a. Any lesion anterior to the equator.
b. Solitary tumours >10 disc diameters behind the equator.
Group IV
a. Multiple tumours, some >10 disc diameters in size.
b. Any lesion extending anteriorly to the ora serrata (the serrated junction between the retina and the ciliary body).
Group V
a. Massive tumours involving over half the retina.
b. Vitreous seeding.
Use of this content is subject to our disclaimer