Although children with DMD (the most common and rapidly progressive muscular dystrophy) can be hypotonic at birth, the classic presentation of DMD is in a toddler with delayed motor milestones, calf hypertrophy, proximal hip girdle muscle weakness, and marked elevation of serum creatine kinase (CK). Elevations in serum muscle enzymes suggest the need for genetic testing to diagnose the condition. As soon as DMD is suspected or diagnosed, patients should be referred to specialty clinics so that the family can receive counseling about current and future treatment options.
The related (but milder) condition Becker muscular dystrophy (BMD), other forms of muscular dystrophy (e.g., myotonic dystrophy, Emery-Dreifuss muscular dystrophy, facioscapulohumeral muscular dystrophy, limb-girdle muscular dystrophies, and congenital muscular dystrophies), and spinal muscular atrophy (SMA), are likewise suspected on the basis of hypotonia, muscle weakness, and/or contractures. Incidence and severity of cardiac and respiratory involvement varies between conditions. All are diagnosed by DNA analysis. Time of presentation differs; some conditions are apparent at birth or soon after (congenital muscular dystrophies, SMA), others may not present until the teenage years or later, and the age of onset of some (e.g., limb-girdle muscular dystrophies) can vary. Similarly, rate of disease progression is variable.[1]Mercuri E, Bönnemann CG, Muntoni F. Muscular dystrophies. Lancet. 2019 Nov 30;394(10213):2025-38.
http://www.ncbi.nlm.nih.gov/pubmed/31789220?tool=bestpractice.com
[3]Mercuri E, Finkel RS, Muntoni F, et al; SMA Care Group. Diagnosis and management of spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018 Feb;28(2):103-15.
https://www.nmd-journal.com/article/S0960-8966(17)31284-1/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/29290580?tool=bestpractice.com
[21]Angelini C, Marozzo R, Pegoraro V. Current and emerging therapies in Becker muscular dystrophy (BMD). Acta Myol. 2019 Sep;38(3):172-9.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6859412
http://www.ncbi.nlm.nih.gov/pubmed/31788661?tool=bestpractice.com
History
When there is a family history of DMD, suspicion is high and diagnosis can almost always be made by DNA analysis. Suspicion would be very much higher for a boy, because the overwhelming majority of patients are male.
For patients with no family history, the diagnosis is often missed at an early stage.[10]Birnkrant DJ, Bushby K, Bann CM, et al; DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018 Mar;17(3):251-67.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5869704
http://www.ncbi.nlm.nih.gov/pubmed/29395989?tool=bestpractice.com
[22]Fox H, Millington L, Mahabeer I, et al. Duchenne muscular dystrophy. BMJ. 2020 Jan 23;368:l7012.
http://www.ncbi.nlm.nih.gov/pubmed/31974125?tool=bestpractice.com
[23]Aartsma-Rus A, Hegde M, Ben-Omran T, et al. Evidence-based consensus and systematic review on reducing the time to diagnosis of Duchenne muscular dystrophy. J Pediatr. 2019 Jan;204:305-13.e14.
https://www.jpeds.com/article/S0022-3476(18)31550-6/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/30579468?tool=bestpractice.com
Motor milestones are delayed, with independent ambulation often not being attained by 18 months. Weakness and clumsiness may be noticed by the parents, and once the child is walking there may be frequent falls, difficulty with climbing stairs, and/or abnormal gait (e.g., toe walking).[10]Birnkrant DJ, Bushby K, Bann CM, et al; DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018 Mar;17(3):251-67.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5869704
http://www.ncbi.nlm.nih.gov/pubmed/29395989?tool=bestpractice.com
[22]Fox H, Millington L, Mahabeer I, et al. Duchenne muscular dystrophy. BMJ. 2020 Jan 23;368:l7012.
http://www.ncbi.nlm.nih.gov/pubmed/31974125?tool=bestpractice.com
[23]Aartsma-Rus A, Hegde M, Ben-Omran T, et al. Evidence-based consensus and systematic review on reducing the time to diagnosis of Duchenne muscular dystrophy. J Pediatr. 2019 Jan;204:305-13.e14.
https://www.jpeds.com/article/S0022-3476(18)31550-6/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/30579468?tool=bestpractice.com
Physical examination
Children with DMD have an imbalance of strength at all lower extremity pivots, with relatively weaker hip extensors, knee extensors, and ankle dorsiflexors. This results in two physical signs.
Gowers sign, where the patient "climbs up his body" to come to stand from a seated position, is characteristic of a child with DMD from the ages of 4 to 7 years.
Musculotendinous contractures are caused by imbalance of lower body strength. The first of these to develop are an increased lumbar lordosis and heel cord contractures.
Gait should be observed; toe walking or a waddling gait is common. Children with DMD also develop calf hypertrophy due to the ongoing regeneration of muscle fibers characteristic of all muscular dystrophies.[10]Birnkrant DJ, Bushby K, Bann CM, et al; DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018 Mar;17(3):251-67.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5869704
http://www.ncbi.nlm.nih.gov/pubmed/29395989?tool=bestpractice.com
[22]Fox H, Millington L, Mahabeer I, et al. Duchenne muscular dystrophy. BMJ. 2020 Jan 23;368:l7012.
http://www.ncbi.nlm.nih.gov/pubmed/31974125?tool=bestpractice.com
This pathokinesiology is also typical for many patients with limb-girdle muscular dystrophy or Emery-Dreifuss muscular dystrophy, and, to a lesser degree, for other neuromuscular diseases. [Figure caption and citation for the preceding image starts]: 7-year-old boy with Duchenne muscular dystrophy standing, maintaining center of gravity behind the hips and anterior to the kneesFrom the collection of John R. Bach, MD, FAAPMR; used with permission [Citation ends].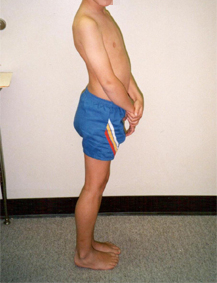
Deep tendon reflexes and muscle tone are diminished in all muscle groups, but all modalities of sensation are normal. If muscle tone and reflexes are increased and sensation is abnormal, an alternative diagnosis needs to be considered.
Learning, attentional, and behavioral difficulties are found in some children with DMD. Other nonmotor signs may include speech and language delay, and faltering growth.[10]Birnkrant DJ, Bushby K, Bann CM, et al; DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018 Mar;17(3):251-67.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5869704
http://www.ncbi.nlm.nih.gov/pubmed/29395989?tool=bestpractice.com
[22]Fox H, Millington L, Mahabeer I, et al. Duchenne muscular dystrophy. BMJ. 2020 Jan 23;368:l7012.
http://www.ncbi.nlm.nih.gov/pubmed/31974125?tool=bestpractice.com
Measurement of creatine kinase (CK)
As soon as muscle disease is suspected, plasma CK levels are measured. Elevated CK levels are characteristic of DMD, and levels of over 20,000 international units/L are not uncommon.[11]Duan D, Goemans N, Takeda S, et al. Duchenne muscular dystrophy. Nat Rev Dis Primers. 2021 Feb 18;7(1):13.
https://www.nature.com/articles/s41572-021-00248-3
http://www.ncbi.nlm.nih.gov/pubmed/33602943?tool=bestpractice.com
[23]Aartsma-Rus A, Hegde M, Ben-Omran T, et al. Evidence-based consensus and systematic review on reducing the time to diagnosis of Duchenne muscular dystrophy. J Pediatr. 2019 Jan;204:305-13.e14.
https://www.jpeds.com/article/S0022-3476(18)31550-6/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/30579468?tool=bestpractice.com
CK levels are typically markedly elevated in most (but not all) other muscular dystrophies, and are usually normal or only slightly elevated in patients with SMA.[1]Mercuri E, Bönnemann CG, Muntoni F. Muscular dystrophies. Lancet. 2019 Nov 30;394(10213):2025-38.
http://www.ncbi.nlm.nih.gov/pubmed/31789220?tool=bestpractice.com
[3]Mercuri E, Finkel RS, Muntoni F, et al; SMA Care Group. Diagnosis and management of spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018 Feb;28(2):103-15.
https://www.nmd-journal.com/article/S0960-8966(17)31284-1/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/29290580?tool=bestpractice.com
Genetic testing
Genetic testing is an essential component for the diagnosis of DMD, BMD, other muscular dystrophies, and SMA.[1]Mercuri E, Bönnemann CG, Muntoni F. Muscular dystrophies. Lancet. 2019 Nov 30;394(10213):2025-38.
http://www.ncbi.nlm.nih.gov/pubmed/31789220?tool=bestpractice.com
[3]Mercuri E, Finkel RS, Muntoni F, et al; SMA Care Group. Diagnosis and management of spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018 Feb;28(2):103-15.
https://www.nmd-journal.com/article/S0960-8966(17)31284-1/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/29290580?tool=bestpractice.com
[10]Birnkrant DJ, Bushby K, Bann CM, et al; DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018 Mar;17(3):251-67.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5869704
http://www.ncbi.nlm.nih.gov/pubmed/29395989?tool=bestpractice.com
[18]Thornton CA. Myotonic dystrophy. Neurol Clin. 2014 Aug;32(3):705-19, viii.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4105852
http://www.ncbi.nlm.nih.gov/pubmed/25037086?tool=bestpractice.com
[21]Angelini C, Marozzo R, Pegoraro V. Current and emerging therapies in Becker muscular dystrophy (BMD). Acta Myol. 2019 Sep;38(3):172-9.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6859412
http://www.ncbi.nlm.nih.gov/pubmed/31788661?tool=bestpractice.com
[23]Aartsma-Rus A, Hegde M, Ben-Omran T, et al. Evidence-based consensus and systematic review on reducing the time to diagnosis of Duchenne muscular dystrophy. J Pediatr. 2019 Jan;204:305-13.e14.
https://www.jpeds.com/article/S0022-3476(18)31550-6/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/30579468?tool=bestpractice.com
When DMD is suspected, testing for deletion or duplication of the dystrophin-encoding DMD gene is carried out first, preferably using multiplex ligation-dependent probe amplification or array comparative genome hybridization. If no mutation is detected, this is followed by detailed genetic sequencing to analyze other, less common mutations associated with DMD.[11]Duan D, Goemans N, Takeda S, et al. Duchenne muscular dystrophy. Nat Rev Dis Primers. 2021 Feb 18;7(1):13.
https://www.nature.com/articles/s41572-021-00248-3
http://www.ncbi.nlm.nih.gov/pubmed/33602943?tool=bestpractice.com
[24]Aartsma-Rus A, Ginjaar IB, Bushby K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet. 2016 Mar;53(3):145-51.
https://jmg.bmj.com/content/53/3/145
http://www.ncbi.nlm.nih.gov/pubmed/26754139?tool=bestpractice.com
If there are existing genetic test results, do not repeat a genetic test unless there is uncertainty about the existing result, e.g., the result is inconsistent with the patient’s clinical presentation or the test methodology has changed.[25]American College of Medical Genetics and Genomics. Five things physicians and patients should question. Choosing Wisely, an initiative of the ABIM Foundation. 2021 [internet publication].
https://web.archive.org/web/20230326143738/https://www.choosingwisely.org/societies/american-college-of-medical-genetics-and-genomics
Genetic testing should also be offered to family members, with appropriate pre- and post-test counseling.[23]Aartsma-Rus A, Hegde M, Ben-Omran T, et al. Evidence-based consensus and systematic review on reducing the time to diagnosis of Duchenne muscular dystrophy. J Pediatr. 2019 Jan;204:305-13.e14.
https://www.jpeds.com/article/S0022-3476(18)31550-6/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/30579468?tool=bestpractice.com
Other investigations
If DNA studies for DMD are negative, electromyography (EMG) and muscle biopsy may be considered. EMG can usually distinguish between neuropathic and myopathic pathology. The EMG in patients with myopathic pathology demonstrates fast-firing, short-duration but polyphasic and decreased amplitude motor units, with early recruitment in the affected muscles. A neurogenic EMG will usually lead the clinician to consider SMA and polyneuropathies.
If the EMG indicates myopathic pathology, muscle biopsy is performed. Muscle biopsy that demonstrates the absence of dystrophin can establish the diagnosis of DMD or Becker muscular dystrophy. Routine histochemistry, immunohistochemistry (assessing dystrophin, sarcoglycans, alpha dystroglycan, merosin, caveolin 3, dysferlinA), and immunoblot testing (for dystrophin, dysferlin, and caplain) are performed on muscle biopsy specimens.[26]Amato AA, Russell JA. Neuromuscular disorders. New York, NY: McGraw-Hill; 2008. Other proteins may be tested for in selected cases (e.g., emerin for Emery-Dreifuss muscular dystrophy).[1]Mercuri E, Bönnemann CG, Muntoni F. Muscular dystrophies. Lancet. 2019 Nov 30;394(10213):2025-38.
http://www.ncbi.nlm.nih.gov/pubmed/31789220?tool=bestpractice.com
Muscle magnetic resonance imaging (MRI) may be helpful in identifying distinctive patterns seen in certain types of muscular dystrophies. It may also be used to assess disease progression.[1]Mercuri E, Bönnemann CG, Muntoni F. Muscular dystrophies. Lancet. 2019 Nov 30;394(10213):2025-38.
http://www.ncbi.nlm.nih.gov/pubmed/31789220?tool=bestpractice.com