Etiology
The cause of essential thrombocythemia (ET) is unknown.
Genetic factors may be involved in the development of ET. Somatic driver mutations in the Janus kinase 2 (JAK2), calreticulin (CALR), or myeloproliferative leukemia virus oncogene (MPL) genes are commonly present in patients with ET and other myeloproliferative neoplasms (primary myelofibrosis, polycythemia vera).[18] These driver mutations can cause abnormal blood cells to grow and proliferate uncontrollably.
The JAK2 V617F, CALR, and MPL mutations are present in approximately 50% to 60%, 25% to 30%, and 3% to 11% of patients with ET, respectively.[18] Approximately 10% to 15% of patients with ET are negative for all three driver mutations (triple‐negative); therefore, absence of these mutations does not exclude the diagnosis.[18] Driver mutation expression is often considered to be mutually exclusive in ET; however, there are reports of patients with coexisting JAK2 V617F and CALR, or JAK2 V617F and MPL, mutations.[19][20]
There are reports of familial ET with autosomal dominant transmission, but specific germline mutations causing familial ET have not been identified.[21][22][23]
As in many hematologic malignancies, environmental and lifestyle factors may be contributory.[24] At present, however, there are no clearly identified etiologic agents or circumstances.
Pathophysiology
ET is a clonal hematopoietic stem cell disorder. It is characterized by proliferation of abnormal megakaryocytes in the bone marrow, resulting in increased production of platelets, causing thrombocytosis. The cause and pathophysiologic process leading to increased platelet production is unclear.
Genetic mutations of the thrombopoietin (THPO) gene have not been implicated in the pathogenesis of ET. However, in a study of a family with hereditary thrombocythemia, mutations in the genes for thrombopoietin or c-MPL were associated with thrombocytosis.[25]
The mechanism by which ET produces either thrombosis or bleeding has not been well defined. Several defects have been described, including hyperaggregation and increased platelet activation (causing thrombosis) and a decrease in aggregation (causing bleeding).
JAK2 V617F mutation is associated with enhanced platelet and leukocyte activation, and a hypercoagulable state, particularly among homozygotes (>50% mutant allele burden) and those who are younger (ages <60 years).[26][27][28][29][30] Patients with JAK2 V617F mutation display higher hemoglobin levels than those without the mutation.[28][31]
Decreases in von Willebrand ristocetin cofactor activity, high-molecular-weight von Willebrand factor multimers, antithrombin III, protein C, and protein S have also been reported.[32][33] Bleeding is more likely in patients with extreme thrombocytosis (i.e., platelet count >1.5 million/microliter) due to an acquired von Willebrand factor deficiency.[Figure caption and citation for the preceding image starts]: Characteristic bone marrow features of essential thrombocythemia in trephine biopsies stained with hematoxylin and eosin. Increase in cellularity (thick arrow) and increase in number of enlarged, mature megakaryocytes with hyperlobulated nuclei (thin arrow)Reproduced with permission from Michiels JJ, de Raeve H, Hebeda K, et al. Leuk Res. 2007 Aug;31(8):1031-8 [Citation ends].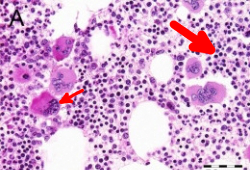
Classification
Use of this content is subject to our disclaimer