Etiology
Osteoporosis is a systemic skeletal disease characterized by a low bone mass and abnormal bone architecture, which leads to compromised bone strength and a consequent increase in bone fragility and risk of fracture.[1][2][3] Bone strength is determined not only by bone mineral density but also by additional factors such as size and shape of the bone, bone turnover, microarchitecture, and bone mineralization. However, no approved tools other than dual-energy x-ray absorptiometry are available as a standard of care method in determining bone strength.
Low bone mass can be the result of low peak bone mass or loss of bone mass with aging. Fragility of bone is not fully explained by low bone mass or bone density (mass/volume). The quality of bone microarchitecture also contributes to bone strength. Bone remodeling and mineralization are important determinants of the quality of bone microarchitecture.
[Figure caption and citation for the preceding image starts]: Scanning electron micrographs showing the structure of L3 vertebra in a woman aged 31 years (top) and in a woman aged 70 years (bottom). Note that many of the plate-like structures have become converted to thin rodsPoole KES, et al. BMJ 2006; 333: 1251; used with permission [Citation ends].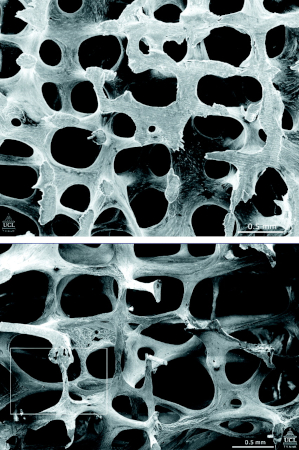
Pathophysiology
Bone is an active tissue that constantly remodels in response to mechanical stresses and hormonal changes.[12]
The bone remodeling process begins with bone resorption by the conversion of quiescent bone surface into bone resorptive surface.[12][13]
By transmitting signals to osteoclasts and osteoblasts on the bone’s surface, osteocytes play a principal role in the initiation of bone remodeling.[14] Osteoclasts resorb bone matrix by first creating a resorption pit. They end their function with apoptosis followed by coupling signals sent to osteoblasts.[15] Osteoblasts then synthesize bone matrix, which undergoes mineralization.
An oversupply or undersupply of osteoclasts or osteoblasts relative to the need for remodeling or cavity repair is fundamental in the pathophysiology of osteoporosis.[12]
Investigation of histomorphometric indices for cancellous bone have demonstrated that men with idiopathic osteoporosis and concomitant hypercalciuria have significantly lower osteoblastic and mineralizing surfaces compared with their normocalciuric counterparts.[16] Epidemiologic studies have shown that osteoporotic fractures occur more frequently in patients with nephrolithiasis and, although the pathophysiology of this connection is unknown, hypercalciuria and hypocitraturia are important risk factors for stone formation.[17][18]
Bone remodeling is regulated by various cytokines, including interleukins 1, 6, and 11, colony-stimulating factors, and calcitropic hormones such as parathyroid hormone, 1,25-dihydroxy vitamin D, calcitonin, and estrogen.[12] It has been shown that members of the tumor necrosis factor (TNF) and TNF-receptor superfamily, receptor activator of nuclear factor-kappa B (RANK), RANK ligand (RANKL), and osteoprotegerin (OPG), play an essential role in osteoclastic bone resorption in postmenopausal osteoporosis.[19] RANKL is expressed by osteoblasts, which interact with the RANK receptors on osteoclasts. Osteoclastic precursor stimulation of RANK facilitates osteoclastic formation, function, and differentiation.[19][20] OPG is secreted by osteoblasts and naturally inhibits the RANKL-induced activation of RANK.
In postmenopausal women with an estrogen deficiency, the overexpression of RANKL activity overrides the natural inhibitory activity of OPG.[20] Osteoporosis is a complex disorder characterized by an imbalance in the bone remodeling process governed by the intricate interactions between various hormonal factors, cytokines, and novel regulatory system RANK/RANKL/OPG. This disequilibrium in bone remodeling leads to lower bone density and bone quality, which culminates in bone fracture.
Use of this content is subject to our disclaimer