Etiology
Gliomas are thought to arise sporadically as a result of the development of somatic oncogenic alterations in central nervous system (CNS) progenitor cells. Only two etiologic risk factors have strong evidence:
Ionizing radiation: the mechanism is related to radiation-induced DNA strand breaks and mutation.[9] Patients with a history of therapeutic radiation to the head are at increased risk of developing a glioblastoma later in life.[10][11]
Rare genetic syndromes that are associated with increased risk of tumor development, including neurofibromatosis type 1, tuberous sclerosis complex, Li-Fraumeni syndrome, and Turcot syndrome.[12][13][14][15]
Other potential risk factors have been investigated, including electromagnetic radiation (including radiation emitted from cellular phones), tobacco, and other environmental factors, but a causal association has not been demonstrated.[16][17]
Autoimmune disorders, asthma, and allergies have been shown to be protecting factors, but the pathophysiology remains unclear.[18][19]
Pathophysiology
The pathophysiology of underlying symptoms in patients with gliomas is thought to be secondary to the infiltration and disruption of normal neural tissue, the pressure or mass effect of the tumor on the surrounding tissue, or the development of vasogenic cerebral edema. These can all correlate with focal neurologic symptoms, or symptoms of high intracranial pressure if the tumor is large or if it obstructs cerebrospinal fluid outflow.
Irritation or infiltration of the underlying cerebral cortex can also lead to the development of seizures.
Rarely, high-grade gliomas may present with acute hemorrhage because of their abnormal vasculature.
Headaches can be produced by high intracranial pressure or by traction on pain-sensitive structures such as the meninges or blood vessels, and are a common symptom (although most headaches in the general population are the result of a primary headache syndrome and do not indicate a primary brain tumor).
Classification
World Health Organization (WHO) classification[1]
The 2021 WHO classification of tumors of the CNS classifies gliomas on the basis of the integration of molecular and histopathologic findings.
Adult-type diffuse gliomas
Astrocytoma, isocitrate dehydrogenase (IDH)-mutant (CNS WHO grades 2, 3, or 4)
Oligodendroglioma, IDH-mutant, and 1p/19q-codeleted (CNS WHO grades 2 or 3)
Glioblastoma, IDH-wildtype (CNS WHO grade 4)
Pediatric-type diffuse low-grade gliomas
Diffuse astrocytoma, MYB- or MYBL1-altered
Angiocentric glioma
Polymorphous low-grade neuroepithelial tumor of the young
Diffuse low-grade glioma, MAPK pathway-altered
Pediatric-type diffuse high-grade gliomas
Diffuse midline glioma, H3 K27-altered
Diffuse hemispheric glioma, H3 G34-mutant
Diffuse pediatric-type high-grade glioma, H3-wildtype and IDH-wildtype
Infant-type hemispheric glioma
Circumscribed astrocytic gliomas
Pilocytic astrocytoma
High-grade astrocytoma with piloid features
Pleomorphic xanthoastrocytoma
Subependymal giant cell astrocytoma
Chordoid glioma
Astroblastoma, MN1-altered
[Figure caption and citation for the preceding image starts]: Simplified classification of diffuse gliomas based on the 2021 WHO classification of CNS tumors. *Astrocytoma, IDH-mutant, can be diagnosed as grade 2, 3, or 4 based on histopathologic grading criteria and cyclin-dependent kinase inhibitor 2A/cyclin-dependent kinase inhibitor 2B (CDKN2A/B) status. ***Oligodendroglioma, IDH-mutant with codeletion of the short arm of chromosome 1 and the long arm of chromosome 19 (1p19q), can be diagnosed as grade 2 or 3 based on histopathologic featuresGritsch et al. Cancer. 2022 Jan 1;128(1):47-58; used with permission [Citation ends].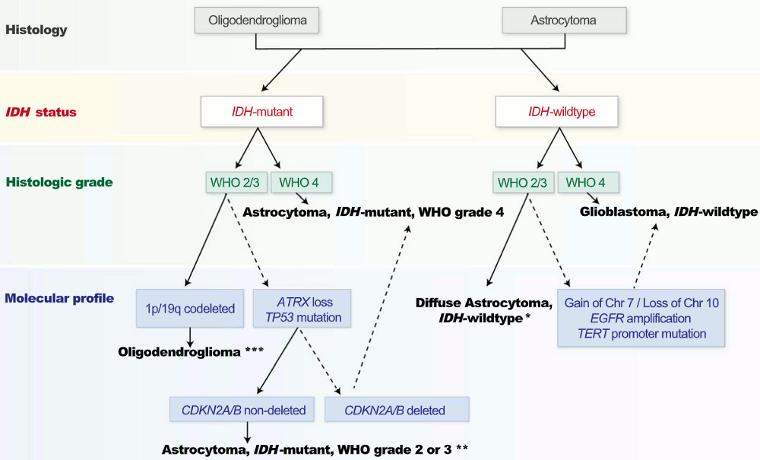
Tumor location
Tumors may be classified by location.
The majority of gliomas are supratentorial, with the exception of diffuse midline gliomas, H3 K27M-altered, and pilocytic astrocytomas, which tend to involve the brainstem/posterior fossa.[2]
The frontal lobe is the most frequent location for gliomas of all grades, followed by the temporal lobe, and less frequently the parietal and occipital lobes.[3]
Intramedullary tumors account for less than 10% of tumors, with astrocytomas and ependymomas accounting for approximately 40% and 60% of these, respectively.[3]
Use of this content is subject to our disclaimer