Approach
Down syndrome (DS) can be definitively diagnosed prenatally via chorionic villus sampling or amniocentesis, or after birth based on the physical findings on the newborn exam leading to karyotype analysis. Chromosomal studies are necessary to both confirm the clinical suspicion and to determine whether the child has standard trisomy 21, a chromosome translocation, or mosaicism.
Physical characteristics
Physical exam within the first 24 hours of life is the most sensitive test for identifying possible DS. Suspicion for DS should prompt confirmation via a chromosomal karyotype test.[17]
The following 10 features of DS are typically present in the neonatal period:[18]
Hypotonia (80% of patients)
Poor Moro reflex (85% of patients)
Hyperflexibility of joints (80% of patients)
Extra skin on back of neck (80% of patients)
Flat facial profile (90% of patients)
Slanted palpebral fissures (80% of patients)
Anomalous auricles (60% of patients)
Hypoplasia of iliac wings (70% of patients)
Short middle phalanx of the fifth finger (60% of patients)
Transverse palmar crease (45% of patients)
General exam in an infant or older child may reveal other findings, although not all features need to be present to suspect the diagnosis.[5][18][Figure caption and citation for the preceding image starts]: Girl with Down syndromeFrom the personal collection of Dr Jeannie Visootsak; photo used with parental consent [Citation ends].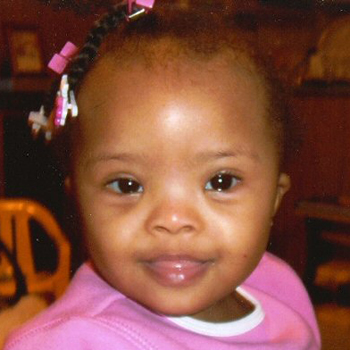 [Figure caption and citation for the preceding image starts]: Boy with mosaic Down syndrome, diagnosed at 18 monthsFrom the personal collection of Dr Jeannie Visootsak; photo used with parental consent [Citation ends].
[Figure caption and citation for the preceding image starts]: Boy with mosaic Down syndrome, diagnosed at 18 monthsFrom the personal collection of Dr Jeannie Visootsak; photo used with parental consent [Citation ends].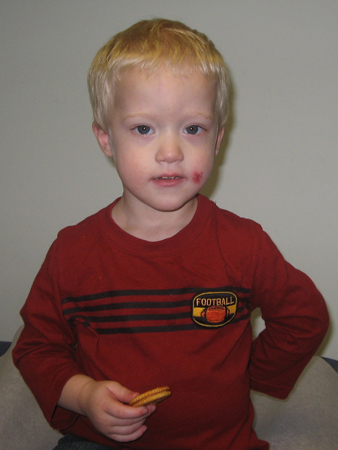 [Figure caption and citation for the preceding image starts]: A 5-year-old boy with Down syndromeFrom the personal collection of Dr Jeannie Visootsak; photo used with parental consent [Citation ends].
[Figure caption and citation for the preceding image starts]: A 5-year-old boy with Down syndromeFrom the personal collection of Dr Jeannie Visootsak; photo used with parental consent [Citation ends].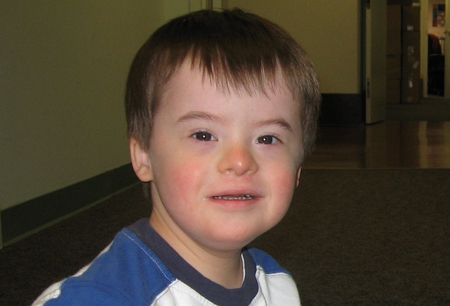
Skull: brachycephaly with a flat occiput
Eyes: epicanthal folds in addition to upslanting palpebral fissures, Brushfield spots of the iris
Nose: short, a low nasal bridge, small nares
Ears: small, may be low-set
Tongue: protruding
Mouth: downturned, small oral cavity
Extremities: short hands, in addition to possible single palmar transverse creases, digital dermatoglyphics, and fifth finger clinodactyly. Wide space between first and second toes, with vertical plantar creases
Diagnosis made postnatally
Specific recommendations have been suggested for delivering the postnatal diagnosis of DS to the family:[17][19][20]
The person delivering the diagnosis should be a clinician with knowledge of DS, preferably an obstetrician and/or a pediatrician (or pediatric subspecialist). Parents often prefer to receive the diagnosis together in a joint meeting with their obstetrician and pediatrician.
Because DS can be identified soon after birth, clinicians should inform parents of their suspicion immediately, even if the diagnosis has not been confirmed by karyotype.
Clinicians should deliver the diagnosis with both parents present in a private hospital room. The infant with DS should be present during the conversation and referred to by name, if one has been chosen.
The clinician should first congratulate the parents on the birth of their child and include information on the positive aspects of DS. Information on DS should be up to date and balanced.
Physicians should provide parents with up-to-date printed material and resources. National Down Syndrome Society Opens in new window National Down Syndrome Congress Opens in new window Global Down Syndrome Foundation Opens in new window Down Syndrome Diagnosis Network Opens in new window
Parents should be offered contact information of local and online support groups, community resources, and other families with children with DS. A directory of local support groups can be located through the National Down Syndrome Society. National Down Syndrome Society Opens in new window
Diagnosis made prenatally
Specific recommendations have been suggested for delivering a prenatal diagnosis of DS to expectant parents:[21][22]
A healthcare professional knowledgeable about DS with specific training in the delivery of sensitive diagnoses should be part of the first conversation. A prenatal diagnosis of DS should be presented in person or at a pre-established time by phone.
Physicians should discuss all the reasons for prenatal investigations: reassurance of a lack of risk, advanced awareness before delivery of the diagnosis of a baby with DS, and the potential for adoption, as well as pregnancy termination.
Results of the prenatal screening should be clearly explained as a risk assessment, not a "positive" or "negative" result. In addition, mothers should first have DS explained to them after the screening test rather than waiting for the results of an amniocentesis or chorionic villus sampling. Mothers should be educated about the benefits and risks associated with subsequent testing procedures.
Test results should be delivered in person, with both parents present, using a sensitive, accurate, and consistent message.
Physicians should provide parents with up-to-date printed material and resources. National Down Syndrome Society Opens in new window National Down Syndrome Congress Opens in new window Global Down Syndrome Foundation Opens in new window Down Syndrome Diagnosis Network Opens in new window
Contact with a local and/or online DS support group should be offered, if desired.
Childhood developmental delay
Many children with DS have global developmental delay. They often have hypotonia, ligamentous laxity, decreased strength, and short arms and legs relative to their trunk length. Motor milestones include rolling between 5.0 and 6.4 months, sitting independently by 14.4 months, crawling between 12.2 and 17.3 months, and walking independently by 54 months.[23][24][25] These milestones may also be impacted by medical conditions such as congenital heart disease (CHD)or hearing loss.
There is typically a language delay that can be further affected by low muscle tone, small mouth with protruding tongue, and open-mouth posture. Difficulties in motor planning and in coordinating rapid movements of the tongue, lips, jaw, and palate affect speech intelligibility. Expressive language is more delayed than receptive language. Other language deficits include development of variability in the number of different words, intelligibility, and mean length of utterance. Overall, expressive language is more delayed than receptive language.[25][26]
Cognitive abilities of a person with DS vary greatly, with challenges in the expressive language domain and strengths in visual-spatial tasks. IQ can range from mild to moderate intellectual disability, between 40 and 72, and may decline with increasing age. The decline in IQ may be explained by a slowing of the developmental course over the childhood years.[27]
Individuals with DS are often social and affectionate, though they may experience challenges that differ from those of their typically developing peers. Some studies quote that 20% to 40% of people with DS have maladaptive behavioral problems. An age-related pattern of behavioral problems may be present: externalizing behavioral problems (opposition, hyperactivity, impulsivity, inattention) are more common during childhood, and internalizing symptoms (shyness, withdrawal, anxiety) are seen in older adolescents.[27]
Chromosomal analysis
Chromosomal studies are indicated to confirm the clinical suspicion and determine whether the karyotype is a standard trisomy 21, a chromosome translocation, or mosaicism. Translocation results require testing the parents to determine future recurrence risks. If there are existing postnatal karyotype results, do not order a duplicate test unless there is uncertainty about the existing result, e.g., the result is inconsistent with the patient’s clinical presentation or the test methodology has changed.[28]
Humans typically have 23 pairs of chromosomes (46 total chromosomes). The first 22 pairs are called autosomes and are similar in males and females. The 23rd pair of chromosomes are called sex chromosomes because they determine sex. The nomenclature of human chromosomes for a male is 46,XY and for a female is 46,XX.[12] Chromosomal variations in DS include:
Standard trisomy 21
An extra chromosome 21 from meiotic nondisjunction or failure of the chromosome pairs to separate during gamete formation is present in about 95% of individuals with DS.[12] Population-based studies show that over 90% of nondisjunction errors leading to trisomy 21 occur in the oocyte and predominantly in maternal meiosis I.[10][11]
The chromosomal karyotype for standard trisomy 21 is indicated as 47,XY,+21 in males and 47,XX,+21 in females.
Robertsonian translocation
About 4% of individuals with DS have 46 chromosomes, one of which is a Robertsonian translocation between 21q and the long arm of one of the other acrocentric chromosomes (typically chromosome 14 or 21).[12] The translocation chromosome replaces one of the acrocentric chromosomes. The karyotype of a person with DS and a Robertsonian translocation between chromosomes 14 and 21 is indicated as 46,XX or XY, der(14;21)(q10;q10),+21.
Translocation DS has a relatively high chance of recurrence in families when one of the parents is a carrier of the translocation. For these reasons, chromosomal karyotyping of the parents is essential to address the potential of recurrence in future pregnancies.
A 21q21q Robertsonian translocation is a chromosome consisting of two chromosome 21 long arms. It is rare. In this case, all gametes of a carrier chromosome contain the 21q21q chromosome, with a double dose of chromosome 21q and a lack of any chromosome 21p genetic material. A carrier of 21q21q translocation has a 100% rate of recurrence in having a child with 21q21q translocation.[12]
Mosaic
About 1% of individuals with DS have mosaic-type DS, with a cell population containing both typical and trisomy 21 karyotype. The phenotype is often believed to be milder than trisomy 21, but cognitive and behavioral profiles vary.
Subsequent investigations immediately after birth
It is important to note specific medical conditions that increase suspicion of DS.[17][29] Some infants with DS may not have the constellation of physical characteristics but present with associated medical conditions.
Evaluation by a pediatric cardiologist, including an echocardiogram, is recommended in all newborns with DS (even in the absence of a murmur). About 50% have CHD.[30]
An abdominal x-ray may be needed in some infants with DS, as they may be born with a gastrointestinal obstruction such as duodenal or anal stenosis or atresia (5% to 12%).[17][31] This should be obtained in any newborn with DS presenting with vomiting, abdominal distention, or delay in stool passage.
Signs of feeding difficulty should be evaluated, especially in the presence of marked hypotonia, poor weight gain, slow feedings or choking with feeds, recurrent or persistent respiratory symptoms, and oxygen desaturation with feeding. Video feeding study and/or nonradiologic videofluoroscopic swallow tests may be indicated if signs are present.
Hearing screen and thyroid tests are required in all newborns in the US. Requirements may vary internationally; please consult local guidance.
A careful ophthalmologic exam should be done in the newborn period because some infants with DS can have glaucoma or congenital cataracts. Cataracts can be assessed via a red reflex test.
Complete blood count with differential should be ordered within 3 days of birth to assess for hematologic abnormalities such as transient abnormal myelopoiesis and polycythemia.
Ongoing monitoring for complications in childhood
The American Academy of Pediatrics (AAP) has developed health supervision guidelines for managing the care of children and adolescents with DS.[17] Children with DS should receive the same preventive health care as any child, with additional monitoring as indicated. For example, children with DS are at increased risk of hearing and visual dysfunction, thyroid problems, and hematologic abnormalities compared to children without DS. Physicians should also continue to monitor the child's developmental, educational, behavioral, and social functioning.
Not all previously recommended routine tests are now considered necessary. For example, the AAP no longer recommends routine screening of asymptomatic children with Down syndrome for celiac disease because there is no evidence of benefit; celiac screening is only recommended in symptomatic patients.[17] Routine cervical spine x-ray for screening of potential atlantoaxial instability is also no longer recommended by the AAP because current evidence does not support this in asymptomatic children with Down syndrome.[17]
See Monitoring for full details.
Use of this content is subject to our disclaimer