Etiology
Congenital adrenal hyperplasia (CAH), of each variety, is an autosomal recessive disorder. The gene encoding the 21-hydroxylase enzyme, CYP21A2 (cytochrome P450 family 21 subfamily A member 2), is mapped to the short arm of chromosome 6 (6p21.3). More than 200 mutations have been described, including point mutations, small deletions, small insertions, and complex rearrangements of the gene.[2] Approximately 95% to 98% of the mutations causing 21-hydroxylase deficiency have been identified through molecular genetic studies of gene rearrangement and point mutations arrays.[7] The genetics of the CYP21A2 gene are complex, in large part due to the existence of a homologous inactive pseudogene - CYP21AP - which is a host to many pathogenic variations. Recombination events, called gene conversions, are operative and lead to incorporation of point mutations, gene deletions, and duplications, as well as development of chimeric genes. Genetic analysis of the parents may need to be performed in certain cases to identify whether two variants exist on the same or different alleles.[8]
Hormonally and clinically defined forms of 21-hydroxylase-deficient CAH are associated with distinct genotypes.[Figure caption and citation for the preceding image starts]: Gene map of two homologs: CYP21A2 (the active gene) and CYP21A1P (the pseudogene). Noncorrespondent bases number less than 90 over a distance of 5.1 kb of DNA. Numbered are the pseudogene base changes that are frequently identified on mutant CYP21A2 genes responsible for 21OHD. Mutations associated with NC21OHD are indicated with black squares (exons 1, 7, 8, and 10)Used with permission from New M. Extensive personal experience: prenatal diagnosis of congenital adrenal hyperplasia in 532 pregnancies. J Clin Endocrinol Metab. 2001:86;5651-5657. Copyright 2001, The Endocrine Society [Citation ends].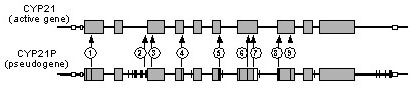 Gene deletions, large gene conversion, and mutations that totally abolish the activity of 21-hydroxylase are associated with classical CAH and are referred to as “severe” mutations. Certain missense mutations, such as p.Val281Leu (V281L) reduce enzymatic activity to approximately 20%, are associated with nonclassical CAH, and are referred to as “mild” mutations.
Gene deletions, large gene conversion, and mutations that totally abolish the activity of 21-hydroxylase are associated with classical CAH and are referred to as “severe” mutations. Certain missense mutations, such as p.Val281Leu (V281L) reduce enzymatic activity to approximately 20%, are associated with nonclassical CAH, and are referred to as “mild” mutations.
The classical form of the disease is seen in patients who carry 2 severe mutations. The nonclassical phenotype is caused by a mild/mild or severe/mild genotype, as is expected in an autosomal recessive disorder. It is not always possible, however, to accurately predict the phenotype on the basis of the genotype. Studies report variable genotype-phenotype concordance, from 50% to over 90%.[9][10]
Pathophysiology
The production of cortisol occurs in the zona fasciculata of the adrenal cortex through 5 enzymatic steps.[Figure caption and citation for the preceding image starts]: Scheme of adrenal corticosteroid synthesisCreated by authors [Citation ends].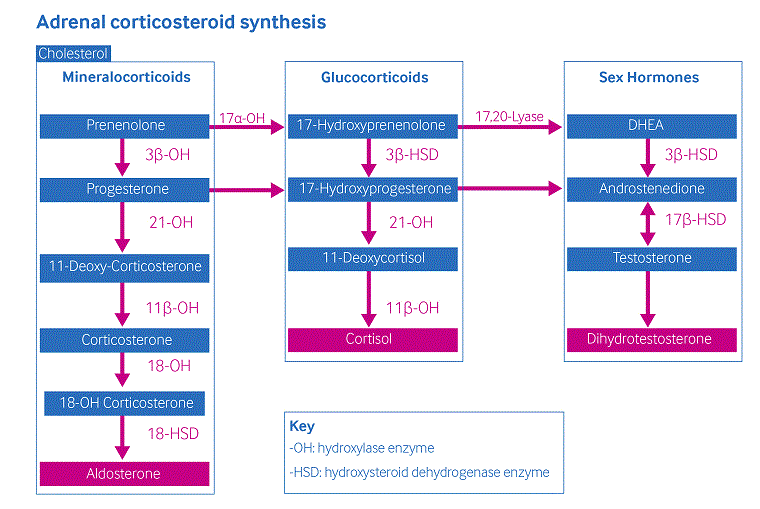
Insufficient cortisol production due to deficiency or decreased activity of the 21-hydroxylase enzyme causes insufficient cortisol production, results in increased production of corticotropin-releasing hormone and adrenocorticotropic hormone (ACTH). High ACTH levels lead to adrenal hyperplasia and over-production of adrenal androgens (e.g., delta-4-androstenedione), which do not require 21-hydroxylation for synthesis. Signs of excessive androgens are found in varied degrees in classical and nonclassical forms of 21-hydroxylase deficiency and are attributable to the severity of the enzyme defect.
In classical CAH, hyperandrogenism is significant and results in virilization of external genitalia in 46,XX fetuses.[2] However, the internal female reproductive tract develops normally, because the adrenal steroidogenic defect does not affect ovarian development postnatal androgen excess may present as virilization (i.e., clitoromegaly in affected females), rapid growth with advanced epiphyseal maturation leading to short adult height, premature development of pubic hair, and the potential for central precocious puberty secondary to peripheral activation of the hypothalamic-pituitary-gonadal (HPG) axis. Excess adrenal androgens may suppress pituitary gonadotropins and thus impair gonadal growth and function.[2]
Gonadal dysfunction usually occurs because the excess adrenal androgens suppress pituitary gonadotropins and thus impair gonadal growth and function.
When the loss of 21-hydroxylase function is severe, adrenal aldosterone secretion is insufficient to stimulate sodium reabsorption by the distal renal tubules, resulting in salt-wasting and cortisol deficiency, in addition to androgen excess.
Classification
Clinical characteristics of CAH caused by 21-hydroxylase deficiency[1]
CAH due to 21-hydroxylase deficiency can be classified as either classical or nonclassical.
Classical CAH has traditionally been subdivided into two forms, salt-wasting and simple virilizing based on relative mineralocorticoid production. There is overlap between the clinical presentation of these two forms and this subdivision is falling out of favor.[2]
Classical CAH: salt-wasting
Most severe form of the disease - characterized by combined cortisol and aldosterone deficiencies and severe adrenal hyperandrogenism
Approximately 75% of classical cases
46,XX infants may present with ambiguous genitalia
46,XY infants (and 46,XX infants with prader 5 genitalia) may present with a salt-wasting crisis in the first 1 to 2 weeks of life as the lack of perceived ambiguity. In these cases, diagnosis may be delayed until the salt-wasting phase. Newborn screening aims to prevent this life-threatening crisis.
May present with atypical genitalia in females
Classical CAH: simple virilizing
Enzyme defect is moderate, affecting cortisol production but not aldosterone
Approximately 25% of classical cases
46,XX infants may present with ambiguous genitalia
46,XY infants may have a delayed diagnosis in the absence of adequate newborn screening but will not present with salt-wasting
Nonclassical CAH
Mild to moderate enzyme deficiency
Present postnatally with signs of hyperandrogenism
Females do not have virilized genitalia at birth
May present in a child as precocious development of axillary hair, pubic hair, penile or clitoral enlargement, acne, or tall stature with an advanced bone age that may eventually result in short stature
Adolescent and adult females may also present with oligomenorrhea, amenorrhea, polycystic ovaries, acne, hirsutism, alopecia, and impaired fertility
Use of this content is subject to our disclaimer