Aetiology
The major source of vitamin D for most humans is exposure to sunlight. Avoiding sun exposure, including heavy sunscreen, season, latitude, and time of day strongly influence cutaneous vitamin D production.[1][58] Increased skin pigmentation results in less effective photoproduction of vitamin D and 25-hydroxyvitamin D.[58][59] Ageing is also considered a risk factor for vitamin D deficiency because the ability of the skin to produce vitamin D decreases with age.[58] The increase in the blood level of vitamin D in young adults aged 20-30 years after exposure to ultraviolet B radiation is at least three-fold higher compared with older adults aged 62-80 years.[58]
Inadequate dietary and supplemental vitamin D intake is another important cause of vitamin D deficiency.[11] Infants who receive their sole nutrition from breast milk, without vitamin D supplementation, are particularly at risk.[60][61] At birth, most extremely preterm infants have biochemical vitamin D deficiency. The risk is reduced or can be prevented with vitamin D supplementation.[62] Patients with intestinal malabsorption syndromes, including coeliac disease, cystic fibrosis, Crohn's disease, Whipple's disease, or short bowel syndrome, as well as those who have undergone gastric bypass surgery, are either unable to absorb vitamin D or they absorb it poorly.[2][63] There is also a possible bidirectional relationship through which poor vitamin D status may negatively impact on the clinical course of the underlying intestinal disease, particularly the inflammatory bowel diseases (Crohn’s disease, ulcerative colitis), as vitamin D has the potential to regulate gut mucosal immunity.[63]
Severe liver failure is associated with vitamin D deficiency.[64] The potential mechanisms for vitamin D deficiency in chronic liver disease include reduced exogenous vitamin D sources (diet, limited exposure to sunlight), the intestinal malabsorption of vitamin D due to cholestasis, the reduced production of vitamin D binding protein, and albumin due to liver injury, impaired hepatic hydroxylation, and increased catabolism of 25-hydroxyvitamin D.[65] When more than 90% of the liver fails, it is incapable of producing enough 25-hydroxyvitamin D.[3]
Vitamin D is fat-soluble: when it is ingested or produced in the skin, most of it is initially incorporated into the body fat. For adults with a BMI >30 the fat sequesters vitamin D; therefore, obesity increases the risk for vitamin D deficiency.[66] Obese adults require two to three times more vitamin D to both treat vitamin D deficiency and prevent recurrence.[2][67]
Using glucocorticoids, antiepileptic medication, highly active antiretroviral therapy, rifampicin, and St John's wort may also result in vitamin D deficiency owing to activation of the steroid and xenobiotic receptors, which results in activation of enzymes that destroy 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D.[68]
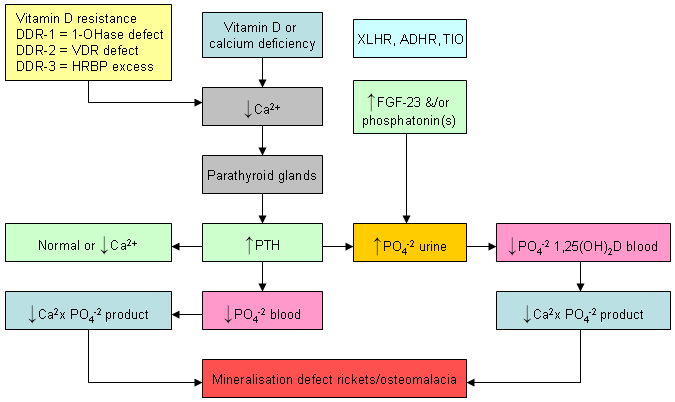
Rarely, a vitamin D deficiency-like syndrome can occur as a result of several inherited disorders that either reduce or prevent the metabolism of vitamin D to 25-hydroxyvitamin D or 25-hydroxyvitamin D to 1,25-dihydroxyvitamin D.[5] These include vitamin D-dependant rickets, X-linked hypophosphataemic rickets, and autosomal-dominant hypophosphataemic rickets.[6][8]
Acquired disorders of vitamin D metabolism, such as chronic kidney disease (CKD), result in retention of phosphate causing an increase in the production of fibroblast growth factor 23 (FGF-23) by osteocytes. The FGF-23 inhibits the production of 1,25-dihydroxyvitamin D and initially results in secondary hyperparathyroidism.[9] Patients with CKD are unable to produce sufficient 1,25-dihydroxyvitamin D to regulate calcium metabolism. In the early stages of CKD (stage 2: GFR 60-89 mL/min/1.73 m²; stage 3A: GFR 45-59 mL/min/1.73 m²), the retention of phosphate causes an increase in serum FGF-23 levels, which in turn decreases the efficiency of the kidneys to produce 1,25-dihydroxyvitamin D. In later stages (stage 3B: GFR 30-44 mL/min/1.73 m²; stage 4: GFR 15-29 mL/min/1.73 m²; and stage 5: GFR <15 mL/min/1.73 m²), the kidneys are unable to produce an adequate amount of 1,25-dihydroxyvitamin D to maintain calcium metabolism. The inability to produce a sufficient amount of 1,25-dihydroxyvitamin D results in secondary hyperparathyroidism and CKD-metabolic bone disease.[69]
Granulomatous disorders (e.g., sarcoidosis, tuberculosis), primary hyperparathyroidism, and hyperthyroidism result in increased metabolism of 25-hydroxyvitamin D to 1,25-dihydroxyvitamin D. The increased production of 1,25-dihydroxyvitamin D results in an increase in the expression and production of the 25-hydroxyvitamin D-24-hydroxylase in the kidneys, which catabolises 25-hydroxyvitamin D to a water-soluble inactive metabolite resulting in reduced levels of 25-hydroxyvitamin D.[2][3] In addition, a benign or malignant tumour may produce an excessive amount of fibroblast growth factor 23 (FGF-23), causing severe hypophosphataemia, low levels of serum 1,25-dihydroxyvitamin D, and consequently oncogenic osteomalacia (also known as tumour-induced osteomalacia).[4]
Pathophysiology
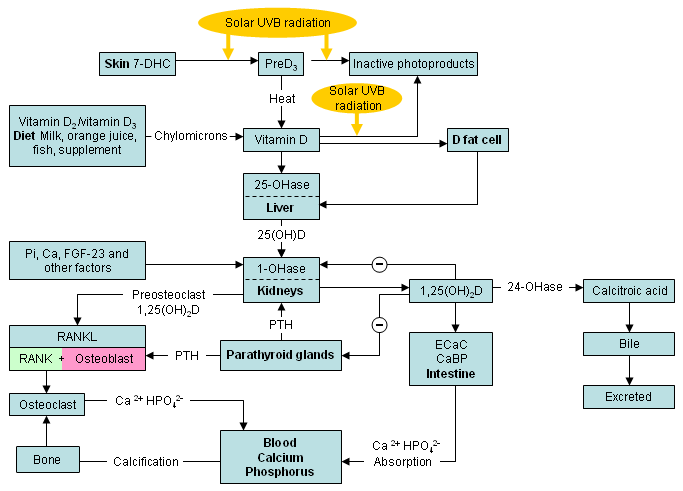
Normal vitamin D metabolism, and actions on calcium and phosphate metabolism
Vitamin D3 obtained from sun-induced production in the skin, and vitamin D2 or vitamin D3 obtained from the diet, are biologically inactive and require hydroxylation in the liver to form 25-hydroxyvitamin D. This is the major circulating form of vitamin D used to determine vitamin D status. 25-hydroxyvitamin D is also inactive and is hydroxylated in the kidneys to 1,25-dihydroxyvitamin D.[1][2][3]
Once formed, 1,25-dihydroxyvitamin D travels to the small intestine and interacts with its nuclear receptor (vitamin D receptor [VDR]) to enhance the efficiency of the intestine to absorb dietary calcium. In a vitamin D-deficient state only about 10% to 15% of dietary calcium is absorbed, whereas vitamin D sufficiency improves intestinal calcium absorption in the range of 30% to 40%.[3] At times, when there is a great increased requirement for calcium (including in teenagers during their growth spurt, pregnancy, and lactation), efficiency increases to as much as 60% to 80%.
1,25-dihydroxyvitamin D also increases phosphate absorption mainly in the jejunum. In a vitamin D-deficient state approximately 60% of dietary phosphate is absorbed, and in a vitamin D-sufficient state this is improved by about 80%.[3] 1,25-dihydroxyvitamin D interacts with its VDR in osteoblasts. This results in an increase in the expression and production of the major non-collagenous protein in the bone, osteocalcin. In addition, it stimulates the expression and production of the receptor activator of nuclear factor kappa-Β ligand (RANKL). This protein binds the RANKL receptor (RANK) on monocytes. This interaction induces these monocytes to coalesce and become a multi-nucleated osteoclast that releases collagenases and HCl to destroy the matrix and release the calcium into the circulation.[3][Figure caption and citation for the preceding image starts]: Production, metabolism, and biological functions of vitamin D on calcium metabolism and bone health (D represents D2 and D3). Abbreviations: CaBP, calbindin; ECaC, epithelial calcium channel; FGF-23, fibroblast growth factor 23; preD3, previtamin D3; RANKL, receptor activator of nuclear factor-kappaB ligand; 1,25(OH)2D, 1,25-dihydroxyvitamin D; 1-OHase, 25-hydroxyvitamin-1-hydroxylase; 7-DHC, 7-dehydrocholesterol; 24-OHase, 1,25-dihydroxyvitamin D-24-hydroxylase; 25-OHase, vitamin D-25-hydroxylase; 25(OH)D, 25-hydroxyvitamin DCreated by M.F. Holick, PhD, MD; used with permission [Citation ends].
 [Figure caption and citation for the preceding image starts]: Chemical structure of vitamin DFrom the collection of M.F. Holick, PhD, MD; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Chemical structure of vitamin DFrom the collection of M.F. Holick, PhD, MD; used with permission [Citation ends].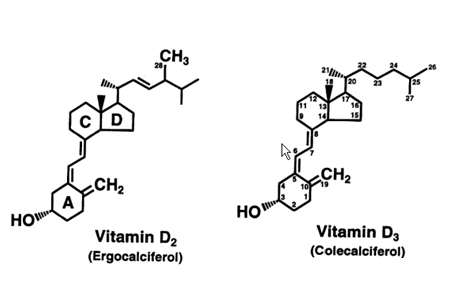
Skeletal action of vitamin D
Children: vitamin D deficiency is the most common cause of rickets.[1][5] Vitamin D deficiency prevents efficient absorption of dietary calcium and phosphorus. The poor absorption of calcium causes a decrease in serum ionised calcium, resulting in secondary hyperparathyroidism. Parathyroid hormone (PTH) decreases phosphorus reabsorption in the kidneys, causing loss of phosphorus into the urine. Therefore, the serum calcium is usually normal in a vitamin D-deficient infant or child. However, the serum phosphorus level is low or low-normal, and therefore there is an inadequate calcium-phosphorus product, causing a defect in the mineralisation of the collagen matrix.
Adults: vitamin D deficiency results in an increase in PTH levels, which in turn increases osteoclastic activity, resulting in the removal of the matrix and mineral from the skeleton. As a result, vitamin D deficiency in adults reduces bone mineral content, leading to osteopenia and osteoporosis. In addition, the secondary hyperparathyroidism results in phosphorus loss in the kidneys, resulting in a normal serum calcium with a low-normal serum phosphorus level. This causes a low calcium-phosphorus product in the blood, which results in the inability of the collagen matrix to be mineralised, leading to osteomalacia. Therefore, for adults, vitamin D deficiency is associated with a normal serum calcium, low-normal or normal serum phosphorus, elevated PTH, and a normal or elevated alkaline phosphatase. The blood level of 25-hydroxyvitamin D is <75 nanomoles/L (<30 nanograms/mL) and the 1,25-dihydroxyvitamin D level is usually normal or elevated.[1][Figure caption and citation for the preceding image starts]: Differential effects of various disorders of calcium, phosphate, bone, and vitamin D metabolism on serum levels of calcium, phosphate, and 1,25-dihydroxyvitamin D. Abbreviations: ADHR, autosomal-dominant hypophosphataemic rickets; FGF-23, fibroblast growth factor 23; HRBP, heterologous ribonuclear binding protein; TIO, tumour-induced osteomalacia; VDR, vitamin D-resistant; XLHR, X-linked hypophosphataemic rickets; 1-OHase, 25-hydroxyvitamin-1-hydroxylaseCreated by M.F. Holick, PhD, MD; used with permission [Citation ends].
 [Figure caption and citation for the preceding image starts]: Bone biopsy of trabecular bone demonstrating (A) increased osteoclastic activity due to secondary hyperparathyroidism; (B) normal; and (C) wide osteoid seams (light pink area), which are classic for osteomalaciaFrom the collection of M.F. Holick, PhD, MD; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Bone biopsy of trabecular bone demonstrating (A) increased osteoclastic activity due to secondary hyperparathyroidism; (B) normal; and (C) wide osteoid seams (light pink area), which are classic for osteomalaciaFrom the collection of M.F. Holick, PhD, MD; used with permission [Citation ends].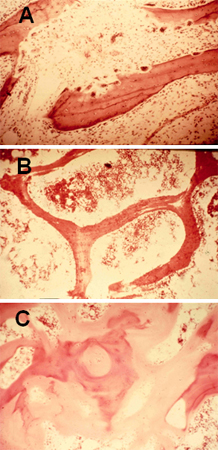
Non-skeletal action of vitamin D
Most tissues and cells in the body have a vitamin D receptor. Furthermore, many of these tissues and cells also have a 25-hydroxyvitamin D-1-hydroxylase capable of converting 25 hydroxyvitamin D to 1,25-dihydroxyvitamin D.[1][3][21]
Vitamin D is a major factor in immune modulation. Activated T and B lymphocytes have a vitamin D receptor, and their immunological activities are regulated by 1,25-dihydroxyvitamin D.[1][21] Macrophages make 1,25-dihydroxyvitamin D for the purpose of inducing the production of cathelicidin, a protein that kills infectious agents, including Mycobacterium tuberculosis.[55] In addition, 1,25-dihydroxyvitamin D manufactured in monocytes or macrophages is most probably released to act locally on activated T and B lymphocytes, which regulate cytokine and immunoglobulin synthesis, respectively. These immunological actions are thought to play a role in reducing the risk for autoimmune diseases and infectious diseases.[21]
The risk of common cancers has been shown to be reduced in the presence of normal 25-hydroxyvitamin D levels. This is thought to be due to the production of 1,25-dihydroxyvitamin D in the breast, colon, prostate, and other tissues, maintaining normal cell proliferation and differentiation. The effect of vitamin D on the reduction of type 2 diabetes is thought to be due to 1,25-dihydroxyvitamin D (produced in the kidneys) entering the circulation and down-regulating renal renin production and stimulating insulin secretion in the pancreas.[1][3][Figure caption and citation for the preceding image starts]: Extra-renal production of 1,25-dihydroxyvitamin D to modulate immune function, cell growth, and insulin production. Abbreviations: AB, activated B lymphocyte; AT, activated T lymphocyte; BS, blood sugar; CD, cathelicidin; LPS, lipopolysaccharide; TLR, toll-like receptor; VDR-RXR, vitamin D receptor-retinoid X receptor; 1,25(OH)2D, 1,25-dihydroxyvitamin D; 1-OHase, 25-hydroxyvitamin-1-hydroxylase; 24-OHase, 1,25-dihydroxyvitamin D-24-hydroxylase; 25(OH)D, 25-hydroxyvitamin DCreated by M.F. Holick, PhD, MD; used with permission [Citation ends].
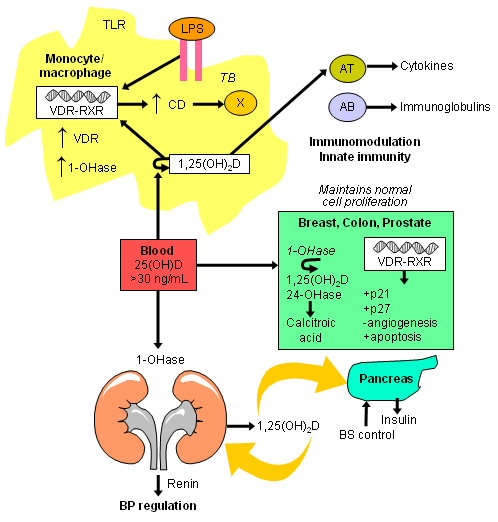 [Figure caption and citation for the preceding image starts]: Schematic representation of paracrine and intracrine function of vitamin D and its metabolites and actions of 1,25-dihydroxyvitamin D on the innate and adaptive immune systems. Abbreviation: 1,25(OH)2D: 1,25-dihydroxyvitamin D; 25(OH)D: 25-hydroxyvitamin D, IFN-Ƴ: interferon- Ƴ; IL: interleukin; MHC: membrane histocompatibility complex, TH1: T helper 1; TH2: T helper 2; TH17: T helper 17; Treg: regulatory T cell, TNF-α: Tumor necrosis factor- α; TLR2: toll-like receptor 2; TLR4: toll-like receptor 4Reproduced with permission from Holick MF, copyright 2020 [Citation ends].
[Figure caption and citation for the preceding image starts]: Schematic representation of paracrine and intracrine function of vitamin D and its metabolites and actions of 1,25-dihydroxyvitamin D on the innate and adaptive immune systems. Abbreviation: 1,25(OH)2D: 1,25-dihydroxyvitamin D; 25(OH)D: 25-hydroxyvitamin D, IFN-Ƴ: interferon- Ƴ; IL: interleukin; MHC: membrane histocompatibility complex, TH1: T helper 1; TH2: T helper 2; TH17: T helper 17; Treg: regulatory T cell, TNF-α: Tumor necrosis factor- α; TLR2: toll-like receptor 2; TLR4: toll-like receptor 4Reproduced with permission from Holick MF, copyright 2020 [Citation ends].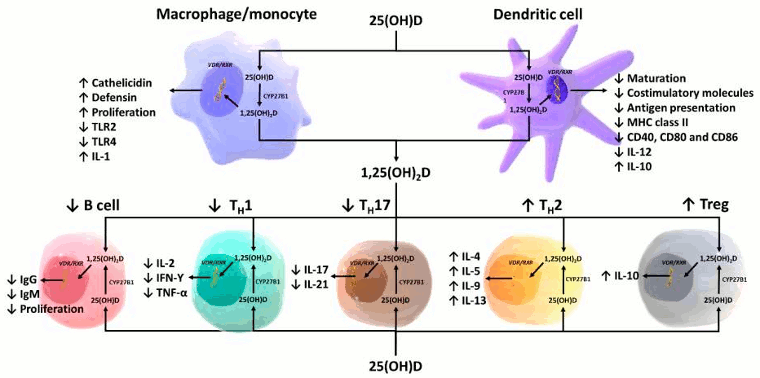
Classification
Aetiological classification[1][2][3]
Acquired:
Nutritional
Related to dietary deficiency, malabsorption, or lack of ultraviolet B light exposure.
Drug-induced
Due to destruction of 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D.
Chronic kidney disease
Due to reduced synthesis of 1,25-dihydroxyvitamin D.
Granulomatous disorders
Macrophages in the granuloma can cause increased metabolism of 25-hydroxyvitamin D to 1,25-dihydroxyvitamin D, resulting in increased expression of the enzyme 24-hydroxylase, which in turn increases the destruction of both 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D, resulting in vitamin D deficiency. The elevated blood levels of 1,25-dihydroxyvitamin D can cause hypercalciuria and hypercalcaemia.
Primary hyperparathyroidism
Due to increased metabolism of 25-hydroxyvitamin D to 1,25-dihydroxyvitamin D. It is, however, the increased production of 1,25-dihydroxyvitamin D that increases the 25-hydroxyvitamin D-24-hydroxylase activity (CYP24A), which in turn metabolises 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D to water-soluble inactive metabolites.
Hyperthyroidism
Due to enhanced metabolism of 25-hydroxyvitamin D.
Oncogenic osteomalacia
Due to the production of fibroblast growth factor 23 (FGF-23) by a benign or malignant tumour; severe hyperphosphaturia and hypophosphataemia result causing osteomalacia. FGF-23 inhibits the renal 25-hydroxyvitamin D-1α-hydroxylase resulting in low levels of serum 1,25-dihydroxyvitamin D.[4]
Inherited:[5]
Vitamin D-dependent rickets type 1
Due to a point mutation of 25-hydroxyvitamin D-1α-hydroxylase or mutations in CYP2R1 that decrease expression or function of the encoded CYP2R1 enzyme, the principal 25-hydroxylase; associated with a low or undetectable level of serum 1,25-dihydroxyvitamin D.[6]
Vitamin D-dependent rickets type 2 (also known as vitamin D-resistant rickets)
Due to mutations of the vitamin D receptor gene (VDR); associated with a very elevated level of serum 1,25-dihydroxyvitamin D.[6]
Vitamin D-dependent rickets type 3
Due to a mutation of the CYP3A4 gene that causes a marked increase in the catabolism of 1,25-dihydroxyvitamin D. Individuals have detectable serum concentrations of the biologically inactive vitamin D3 (generated through sun exposure or obtained from diet) but low serum concentrations the vitamin D metabolites 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D.[7]
X-linked hypophosphataemic rickets
Due to a mutation of the PHEX gene resulting in a decrease in the metabolism of FGF-23, causing severe hyperphosphaturia and hypophosphataemia, leading to severe rickets. Level of serum 1,25-dihydroxyvitamin D is inappropriately low for the presence of hypophosphataemia.[8]
Autosomal-dominant hypophosphataemic rickets
Due to a mutation in the FGF-23 gene at a location where the resultant protein is less sensitive to proteolysis, leading to markedly elevated levels of FGF-23 causing severe hyperphosphaturia and hypophosphataemia, resulting in severe rickets. Level of serum 1,25-dihydroxyvitamin D is inappropriately low for the presence of hypophosphataemia.[8]
Use of this content is subject to our disclaimer