Etiology
EDS is caused by one or more genetic aberrations affecting genes encoding for, or modifying, connective tissue proteins, such as collagen and matrix proteins (e.g., tenascin). Specific genetic aberrations lead to specific risks, such as severe skin pathology in the classical type and vascular collapse due to arteriovenous rupture in the vascular subtype.[12] In some cases, the gene product modifies a connective tissue protein (e.g., kyphoscoliotic type and dermatosparaxis EDS).
The exact genetic variant is not always known and the inheritance pattern is variable.
In cases of hypermobile EDS, the pattern of inheritance is autosomal dominant, so that 50% of offspring of an affected person would be expected to inherit the mutated gene and develop the phenotype.[13] Classical, vascular, arthrochalasia, and periodontal subtypes are also autosomal dominant in nature, whereas the other subtypes are inherited in an autosomal recessive manner.
Studies show that the heritability factor (the proportion of phenotypic variation in a population that is attributable to genetic variation among people) of joint hypermobility is >70%.[14]
Most affected people do not develop symptoms at all, or they develop only minor symptoms during their lifetime.
Pathophysiology
The genetic aberration in genes encoding connective tissue proteins gives rise to a biochemical abnormality, which in turn results in a biomechanical disorder. This has three effects:
Ligament laxity and the resulting hypermobility and enhanced flexibility that is a positive selection factor in performing arts such as dance, gymnastics, and music performance.
Inherent fragility of connective tissues, predisposing the person to injury and increasing vulnerability to the effects of injury.
Impaired healing, which is often delayed and may be incomplete.
Chronic pain and fatigue may ensue, alongside cardiovascular and physical deconditioning.
Classification
The 2017 international classification of the Ehlers-Danlos syndromes[5]
Clinical classification that recognizes 13 subtypes.[Figure caption and citation for the preceding image starts]: The 2017 international classification of the Ehlers-Danlos syndromesAdapted with permission from Malfait F, et al. Am J Med Genet C Semin Med Genet. 2017 Mar;175(1):8-26 [Citation ends].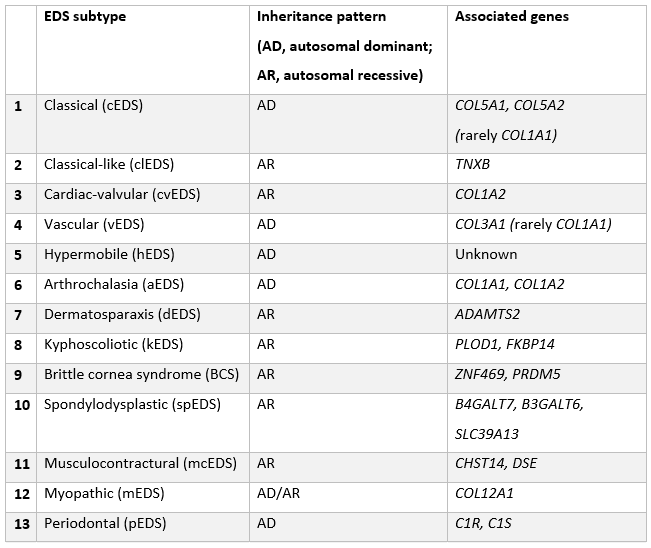
A framework for the classification of joint hypermobility and related conditions[4]
The accepted terminology is joint hypermobility (JH) or hypermobility spectrum disorder (HSD) or hEDS.
Names such as EDS type III, EDS hypermobility type, hypermobility syndrome, joint hypermobility syndrome, and benign joint hypermobility syndrome are now considered outdated.[4]
Use of this content is subject to our disclaimer