History and physical exam (including slit-lamp ophthalmic examination with pupil dilation) in conjunction with imaging of the aortic root and the ascending, descending, and abdominal aorta (echo, computed tomography [CT], magnetic resonance imaging [MRI]) are usually sufficient for diagnosis.[1]Pyeritz AE. The Marfan syndrome. Annu Rev Med. 2000;51:481-510.
http://www.ncbi.nlm.nih.gov/pubmed/10774478?tool=bestpractice.com
Use of diagnostic criteria
The diagnosis of Marfan syndrome is clinically based using well-defined criteria, namely the revised Ghent diagnostic criteria.[2]Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010 Jul;47(7):476-85.
http://www.ncbi.nlm.nih.gov/pubmed/20591885?tool=bestpractice.com
[3]Tinkle BT, Lacro RV, Burke LW, et al. Health supervision for children and adolescents with Marfan syndrome. Pediatrics. 2023 Apr 1;151(4):e2023061450.
http://www.ncbi.nlm.nih.gov/pubmed/36938616?tool=bestpractice.com
This revised diagnostic criteria put more weight on cardiovascular manifestations of the disorder, compared with previously used criteria, with aortic root aneurysm and ectopia lentis now becoming cardinal features.[2]Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010 Jul;47(7):476-85.
http://www.ncbi.nlm.nih.gov/pubmed/20591885?tool=bestpractice.com
[3]Tinkle BT, Lacro RV, Burke LW, et al. Health supervision for children and adolescents with Marfan syndrome. Pediatrics. 2023 Apr 1;151(4):e2023061450.
http://www.ncbi.nlm.nih.gov/pubmed/36938616?tool=bestpractice.com
Identification of risk factors
Risk factors include the presence of a family history of Marfan syndrome, or of aortic dissection or aneurysm. There is also a weak association with high paternal age.[4]Murdoch JL, Walker BA, McKusick VA. Parental age effects on the occurrence of new mutations for the Marfan syndrome. Ann Hum Genet. 1972 Mar;35(3):331-6.
http://www.ncbi.nlm.nih.gov/pubmed/5072691?tool=bestpractice.com
Other historical considerations
There may be a family history of myopia, astigmatism, strabismus, amblyopia, premature cataract or other lens abnormalities, glaucoma, retinal detachment, dental extraction or braces for dental crowding, hernias, or spontaneous pneumothorax. Patients may have a history of joint pain or low back ache.[1]Pyeritz AE. The Marfan syndrome. Annu Rev Med. 2000;51:481-510.
http://www.ncbi.nlm.nih.gov/pubmed/10774478?tool=bestpractice.com
Physical examination
Tall stature, wide arm span, high level of pubic bone, high arched palate, arachnodactyly, positive wrist and thumb sign, pectus excavatum, pectus carinatum, scoliosis, striae (other than from pregnancy/weight change), flat feet, thick eye glasses for myopia, hernias, aortic or mitral valve murmur may be present.[2]Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010 Jul;47(7):476-85.
http://www.ncbi.nlm.nih.gov/pubmed/20591885?tool=bestpractice.com
[3]Tinkle BT, Lacro RV, Burke LW, et al. Health supervision for children and adolescents with Marfan syndrome. Pediatrics. 2023 Apr 1;151(4):e2023061450.
http://www.ncbi.nlm.nih.gov/pubmed/36938616?tool=bestpractice.com
Spontaneous pneumothorax or emphysematous bullae may present as dyspnea.[3]Tinkle BT, Lacro RV, Burke LW, et al. Health supervision for children and adolescents with Marfan syndrome. Pediatrics. 2023 Apr 1;151(4):e2023061450.
http://www.ncbi.nlm.nih.gov/pubmed/36938616?tool=bestpractice.com
Skeletal abnormality may result in other pulmonary complications also presenting as dyspnea.
There may be signs of heart failure due to valve disease or cardiomyopathy.[18]Alpendurada F, Wong J, Kiotsekoglou A, et al. Evidence for Marfan cardiomyopathy. Eur J Heart Fail. 2010 Oct;12(10):1085-91.
http://www.ncbi.nlm.nih.gov/pubmed/20861133?tool=bestpractice.com
A complete ophthalmic exam, including fundus examination with pupil dilation, is recommended in all patients. There may be signs of lens subluxation or dislocation, cataract, glaucoma, or retinal detachment.[3]Tinkle BT, Lacro RV, Burke LW, et al. Health supervision for children and adolescents with Marfan syndrome. Pediatrics. 2023 Apr 1;151(4):e2023061450.
http://www.ncbi.nlm.nih.gov/pubmed/36938616?tool=bestpractice.com
It is possible that the patient may present with signs and symptoms of acute aortic dissection or rupture. This presentation is covered in the complications section.[Figure caption and citation for the preceding image starts]: Positive thumb signFrom the collection of LG Svensson, E Mendrinos, C Pournaras [Citation ends].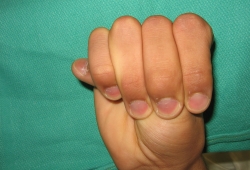
Initial investigations
Echocardiography is used initially for aortic root imaging and if not clearly determined CT or MRI may be used.[3]Tinkle BT, Lacro RV, Burke LW, et al. Health supervision for children and adolescents with Marfan syndrome. Pediatrics. 2023 Apr 1;151(4):e2023061450.
http://www.ncbi.nlm.nih.gov/pubmed/36938616?tool=bestpractice.com
[19]Isselbacher EM, Preventza O, Black JH 3rd, et al. 2022 ACC/AHA guideline for the diagnosis and management of aortic disease: a report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation. 2022 Dec 13;146(24):e334-482.
https://www.ahajournals.org/doi/full/10.1161/CIR.0000000000001106
http://www.ncbi.nlm.nih.gov/pubmed/36322642?tool=bestpractice.com
Subsequent investigations
Genetic screening for mutations in the fibrillin-1 (FBN1) gene can confirm the diagnosis if in doubt.[3]Tinkle BT, Lacro RV, Burke LW, et al. Health supervision for children and adolescents with Marfan syndrome. Pediatrics. 2023 Apr 1;151(4):e2023061450.
http://www.ncbi.nlm.nih.gov/pubmed/36938616?tool=bestpractice.com
In the absence of lens dislocation, genetic testing should be considered to exclude the possibility of Loeys-Dietz syndrome.[19]Isselbacher EM, Preventza O, Black JH 3rd, et al. 2022 ACC/AHA guideline for the diagnosis and management of aortic disease: a report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation. 2022 Dec 13;146(24):e334-482.
https://www.ahajournals.org/doi/full/10.1161/CIR.0000000000001106
http://www.ncbi.nlm.nih.gov/pubmed/36322642?tool=bestpractice.com
Once detected, the mutation can be used to screen other relatives, and used for prenatal diagnosis and preimplantation genetic diagnosis. This test is more specific than MRI for dural ectasia, which can also be found in Ehlers-Danlos syndrome. If there are existing genetic test results, do not order a duplicate test unless there is uncertainty about the existing result, e.g., the result is inconsistent with the patient’s clinical presentation or the test methodology has changed.[20]American College of Medical Genetics and Genomics. Five things physicians and patients should question. Choosing Wisely, an initiative of the ABIM Foundation. 2021 [internet publication].
https://web.archive.org/web/20230326143738/https://www.choosingwisely.org/societies/american-college-of-medical-genetics-and-genomics
Abdominal ultrasound, CT, and MRI are used for visualization of the descending aorta.[19]Isselbacher EM, Preventza O, Black JH 3rd, et al. 2022 ACC/AHA guideline for the diagnosis and management of aortic disease: a report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation. 2022 Dec 13;146(24):e334-482.
https://www.ahajournals.org/doi/full/10.1161/CIR.0000000000001106
http://www.ncbi.nlm.nih.gov/pubmed/36322642?tool=bestpractice.com
Chest x-ray may be performed to exclude the presence of a pneumothorax and may reveal emphysematous bullae.
Lower spine CT scan or MRI can be performed to exclude dural ectasia, where upright imaging may be preferred.[3]Tinkle BT, Lacro RV, Burke LW, et al. Health supervision for children and adolescents with Marfan syndrome. Pediatrics. 2023 Apr 1;151(4):e2023061450.
http://www.ncbi.nlm.nih.gov/pubmed/36938616?tool=bestpractice.com
This is a widening of the dural membrane surrounding the spinal cord and is a recognized complication of Marfan syndrome. MRI is particularly useful for follow-up investigations to avoid accumulative radiation, assessing aortic size and extent of any dural ectasia.
Plasma homocysteine levels help in unclear cases to differentiate homocystinuria.