Patient presentation can be very helpful in determining if symptoms are consistent with Creutzfeldt-Jakob disease (CJD) or if they can be attributed to another condition. An exclusionary approach should be used when diagnosing patients, although brain diffusion-weighted imaging (DWI) and attenuated diffusion coefficient map (ADC) MRI findings have high diagnostic utility.[65]Young GS, Geschwind MD, Fischbein NJ, et al. Diffusion-weighted and fluid-attenuated inversion recovery imaging in Creutzfeldt-Jakob disease: high sensitivity and specificity for diagnosis. AJNR Am J Neuroradiol. 2005 Jun-Jul;26(6):1551-62.
http://www.ajnr.org/content/26/6/1551.full
http://www.ncbi.nlm.nih.gov/pubmed/15956529?tool=bestpractice.com
[66]Zerr I, Kallenberg K, Summers DM, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. 2009 Oct;132(Pt 10):2659-68.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2759336/?tool=pubmed
http://www.ncbi.nlm.nih.gov/pubmed/19773352?tool=bestpractice.com
[67]Vitali P, Maccagnano E, Caverzasi E, et al. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology. 2011 May 17;76(20):1711-9.
http://www.ncbi.nlm.nih.gov/pubmed/21471469?tool=bestpractice.com
Real-time quaking-induced conversion (RT-QuIC) of the CSF also has high diagnostic value.[68]Groveman BR, Orrú CD1, Hughson AG, et al. Extended and direct evaluation of RT-QuIC assays for Creutzfeldt-Jakob disease diagnosis. Ann Clin Transl Neurol. 2016 Dec 27;4(2):139-144.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5288466
http://www.ncbi.nlm.nih.gov/pubmed/28168213?tool=bestpractice.com
[69]Hermann P, Appleby B, Brandel JP, et al. Biomarkers and diagnostic guidelines for sporadic Creutzfeldt-Jakob disease. Lancet Neurol. 2021 Mar;20(3):235-246.
https://www.doi.org/10.1016/S1474-4422(20)30477-4
http://www.ncbi.nlm.nih.gov/pubmed/33609480?tool=bestpractice.com
[70]Centers for Disease Control and Prevention. CDC’s Diagnostic Criteria for Creutzfeldt-Jakob Disease (CJD). 2018 [internet publication].
https://www.cdc.gov/prions/cjd/diagnostic-criteria.html
History
The onset of sporadic CJD (sCJD) is subacute, but overall progress is quite rapid relative to other slower-developing dementias such as Alzheimer disease. Potential risk factors should be noted including certain medical procedures that the patient may have undergone (iatrogenic CJD) and overseas trip details (dates, duration, consumption of beef), particularly in the UK for variant CJD (vCJD).
Upward of 60% of CJD patients found to carry a mutation in the prion protein gene (PRNP) were not known to have a family history of prion disease. Capturing a detailed family tree is important but it is recommended that all patients be tested for gene mutation even if there is no family history. The family history should be evaluated for dementia, other neurologic conditions, and psychiatric disorders, as genetic forms of prion disease are often misdiagnosed as other conditions.
Neurologic and physical exam
The clinical presentation of prion disease probably depends on the regions of the brain in which the prion is accumulating. Symptoms can mimic other neurologic or psychiatric conditions. Patients suspected of having prion disease should be immediately referred to a neurologist. A detailed neurologic exam is vital. Features found on exam will help direct the exclusionary workup.
Most commonly, sCJD occurs in the seventh decade of life (patients in their 60s), and typically presents with the following.
Cognitive complaints, lack of coordination, behavioral and/or visual changes.[71]Geschwind MD, Jay C. Assessment of rapidly progressive dementias. Concise review related to chapter 362: Alzheimer disease and other primary dementias. In: Braunwald E, Harrison TR, Fauci AS, et al, eds. Harrison's principles of internal medicine. New York, NY: McGraw-Hill; 2001:2391-2398.
In a study of more than 100 CJD cases, cognitive problems were the most common first symptom.[71]Geschwind MD, Jay C. Assessment of rapidly progressive dementias. Concise review related to chapter 362: Alzheimer disease and other primary dementias. In: Braunwald E, Harrison TR, Fauci AS, et al, eds. Harrison's principles of internal medicine. New York, NY: McGraw-Hill; 2001:2391-2398. Patients frequently experience memory loss, aphasia, and difficulty with executive functioning (e.g., organizing, planning, and multitasking).
Cerebellar, other motor, and behavioral symptoms were the next most common symptoms. Motor features include parkinsonism, myoclonus, and limb and/or gait ataxia. Involvement of the frontal lobes or frontal subcortical connections can affect behavior causing agitation, depression, and other psychiatric features.[72]Rabinovici GD, Wang PN, Levin J, et al. First symptom in sporadic Creutzfeldt-Jakob disease. Neurology. 2006 Jan 24;66(2):286-7.
http://www.ncbi.nlm.nih.gov/pubmed/16434680?tool=bestpractice.com
Some patients may also describe having nonspecific or constitutional symptoms such as vertigo, headaches, and dizziness that may precede the disease by weeks or even months.[73]Prusiner SB, Bosque PJ. Prion diseases. In: Braunwald E, Harrison TR, Fauci AS, et al, eds. Harrison's principles of internal medicine. New York, NY: McGraw-Hill; 2001:2486-2491.
Visual symptoms are generally less common, but may include diplopia, hallucinations, and other visual distortions.[72]Rabinovici GD, Wang PN, Levin J, et al. First symptom in sporadic Creutzfeldt-Jakob disease. Neurology. 2006 Jan 24;66(2):286-7.
http://www.ncbi.nlm.nih.gov/pubmed/16434680?tool=bestpractice.com
vCJD presents quite differently from the sporadic form.
It typically affects young adults and teens.
In most patients, the first symptoms are psychiatric, including profound depression and mild cognitive impairment.
Later on in the course, patients develop dementia, ataxia, painful sensory symptoms, and/or a movement disorder.[11]Will RG, Zeidler M, Stewart GE, et al. Diagnosis of new variant Creutzfeldt-Jakob disease. Ann Neurol. 2000 May;47(5):575-82.
http://www.ncbi.nlm.nih.gov/pubmed/10805327?tool=bestpractice.com
[21]Will R. Variant Creutzfeldt-Jakob disease. Folia Neuropathol. 2004;42(suppl A):77-83.
http://www.ncbi.nlm.nih.gov/pubmed/15449462?tool=bestpractice.com
[71]Geschwind MD, Jay C. Assessment of rapidly progressive dementias. Concise review related to chapter 362: Alzheimer disease and other primary dementias. In: Braunwald E, Harrison TR, Fauci AS, et al, eds. Harrison's principles of internal medicine. New York, NY: McGraw-Hill; 2001:2391-2398.
Genetic CJD is due to more than 40 different mutations in the prion gene (PRNP) and may be further subdivided into familial CJD, Gerstmann-Straussler-Scheinker (GSS), and fatal familial insomnia (FFI) based on clinical and pathologic findings.
Familial CJD can have a longer, slower course than GSS or FFI.
The clinical course of GSS is typically longer and slower than that of sCJD - often for a few years and as long as a decade. Parkinsonism or ataxia may be the presenting signs of GSS, which can be misdiagnosed as other slower neurodegenerative conditions such as atypical parkinsonian dementias, Parkinson disease, and multiple system atrophy.[74]Collins S, McLean CA, Masters CL. Gerstmann-Sträussler-Scheinker syndrome,fatal familial insomnia, and kuru: a review of these less common human transmissible spongiform encephalopathies. J Clin Neurosci. 2001 Sep;8(5):387-97.
http://www.ncbi.nlm.nih.gov/pubmed/11535002?tool=bestpractice.com
FFI typically presents as a syndrome of insomnia and dysautonomia. Ataxia or cerebellar incoordination can occur. Dementia occurs later in the course of the disease.[75]Gambetti P, Kong Q, Zou W, et al. Sporadic and familial CJD: classification and characterisation. Br Med Bull. 2003;66:213-39.
http://www.ncbi.nlm.nih.gov/pubmed/14522861?tool=bestpractice.com
MRI
MRI should be ordered as soon as a rapidly progressive dementia is suspected. Findings are especially valuable in the diagnosis of prion disease, and have a high sensitivity and specificity when using fluid-attenuated inversion recovery (FLAIR) and particularly diffusion-weighted imaging (DWI) or attenuated diffusion coefficient map (ADC) imaging.[66]Zerr I, Kallenberg K, Summers DM, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. 2009 Oct;132(Pt 10):2659-68.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2759336/?tool=pubmed
http://www.ncbi.nlm.nih.gov/pubmed/19773352?tool=bestpractice.com
[67]Vitali P, Maccagnano E, Caverzasi E, et al. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology. 2011 May 17;76(20):1711-9.
http://www.ncbi.nlm.nih.gov/pubmed/21471469?tool=bestpractice.com
[76]American College of Radiology. ACR appropriateness criteria: movement disorders and neurodegenerative diseases. 2019 [internet publication].
https://acsearch.acr.org/docs/3111293/Narrative
[Figure caption and citation for the preceding image starts]: Changes in the basal ganglia seen in Creutzfeldt-Jakob disease. (A) Fluid-attenuated inversion recovery MRI and (B) diffusion-weighted MRI of the same patient demonstrate bilateral basal ganglia hyperintensities (arrows). There is also mild bilateral medial thalamus and pulvinar hyperintensityFrom the personal collection of Dr M. Geschwind [Citation ends].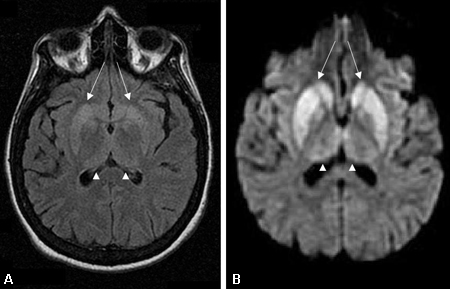 [Figure caption and citation for the preceding image starts]: Bilateral medial thalamus and pulvinar hyperintensity (arrowheads) on (A) fluid-attenuated inversion recovery and (B) diffusion-weighted MRI in a patient with Creutzfeldt-Jakob. This patient also has significant basal ganglia hyperintensity on both sequences (arrows)From the personal collection of Dr M. Geschwind [Citation ends].
[Figure caption and citation for the preceding image starts]: Bilateral medial thalamus and pulvinar hyperintensity (arrowheads) on (A) fluid-attenuated inversion recovery and (B) diffusion-weighted MRI in a patient with Creutzfeldt-Jakob. This patient also has significant basal ganglia hyperintensity on both sequences (arrows)From the personal collection of Dr M. Geschwind [Citation ends]. [Figure caption and citation for the preceding image starts]: Diffuse cortical ribboning (arrows) seen on (A) diffusion-weighted imaging (DWI) and less so on (B) fluid-attenuated inversion recovery (FLAIR) MRI. Both sequences show cerebral cortex gyral hyperintensitiesFrom the personal collection of Dr M. Geschwind [Citation ends].
[Figure caption and citation for the preceding image starts]: Diffuse cortical ribboning (arrows) seen on (A) diffusion-weighted imaging (DWI) and less so on (B) fluid-attenuated inversion recovery (FLAIR) MRI. Both sequences show cerebral cortex gyral hyperintensitiesFrom the personal collection of Dr M. Geschwind [Citation ends].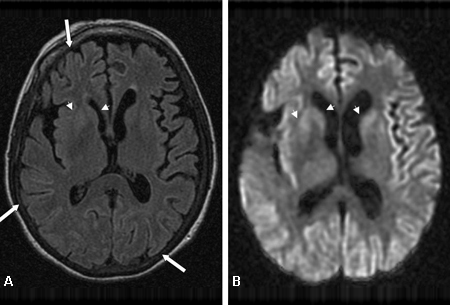
FLAIR and DWI MRI demonstrate hyperintensity in the cerebral cortex gray matter gyri, basal ganglia (caudate and putamen), and, less commonly, thalamus.[67]Vitali P, Maccagnano E, Caverzasi E, et al. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology. 2011 May 17;76(20):1711-9.
http://www.ncbi.nlm.nih.gov/pubmed/21471469?tool=bestpractice.com
[77]Shiga Y, Miyazawa K, Sato S, et al. Diffusion-weighted MRI abnormalities as an early diagnostic marker for Creutzfeldt-Jakob disease. Neurology. 2004 Aug 10;63(3):443-9.
http://www.ncbi.nlm.nih.gov/pubmed/15304574?tool=bestpractice.com
On T1 images with gadolinium, CJD generally does not show contrast enhancement or white matter intensity abnormalities. If these are found, other diagnoses should be investigated.[78]Finkenstaedt M, Szudra A, Zerr I, et al. MR imaging of Creutzfeldt-Jakob disease. Radiology. 1996 Jun;199(3):793-8.
http://www.ncbi.nlm.nih.gov/pubmed/8638007?tool=bestpractice.com
[79]Collie DA, Sellar RJ, Zeidler M, et al. MRI of Creuztfeldt-Jakob disease: imaging features and recommended MRI protocol. Clin Radiol. 2001 Sep;56(9):726-39.
http://www.ncbi.nlm.nih.gov/pubmed/11585394?tool=bestpractice.com
Some CJD patients have T1 hyperintensity in the globus pallidus.
The pulvinar sign, a term referencing bilateral pulvinar hyperintensity, can be seen in vCJD, and when both the pulvinar and medial thalamic show signal intensity, vCJD should be suspected.[71]Geschwind MD, Jay C. Assessment of rapidly progressive dementias. Concise review related to chapter 362: Alzheimer disease and other primary dementias. In: Braunwald E, Harrison TR, Fauci AS, et al, eds. Harrison's principles of internal medicine. New York, NY: McGraw-Hill; 2001:2391-2398.[80]Keohane C. Pulvinar sign on MRI images in variant Creutzfeldt-Jakob disease. Lancet. 2000 Apr 22;355(9213):1384.
http://www.ncbi.nlm.nih.gov/pubmed/10791518?tool=bestpractice.com
[81]Zeidler M, Sellar RJ, Collie DA, et al. The pulvinar sign on magnetic resonance imaging in variant Creutzfeldt- Jakob disease. Lancet. 2000 Apr 22;355(9213):1412-8.
http://www.ncbi.nlm.nih.gov/pubmed/10791525?tool=bestpractice.com
The ADC and DWI abnormalities found on MRI in sCJD and vCJD generally have not been reported in other similar dementias and are strongly suggestive of CJD.[67]Vitali P, Maccagnano E, Caverzasi E, et al. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology. 2011 May 17;76(20):1711-9.
http://www.ncbi.nlm.nih.gov/pubmed/21471469?tool=bestpractice.com
EEG
EEG is routine. Findings can include generalized slowing, focal or diffuse, and periodic sharp-wave complexes. These abnormalities, although moderately specific, are only about 60% sensitive and may not appear until the later stages of the disease.[77]Shiga Y, Miyazawa K, Sato S, et al. Diffusion-weighted MRI abnormalities as an early diagnostic marker for Creutzfeldt-Jakob disease. Neurology. 2004 Aug 10;63(3):443-9.
http://www.ncbi.nlm.nih.gov/pubmed/15304574?tool=bestpractice.com
[82]Steinhoff BJ, Zerr I, Glatting M, et al. Diagnostic value of periodic complexes in Creutzfeldt-Jakob disease. Ann Neurol. 2004 Nov;56(5):702-8.
http://www.ncbi.nlm.nih.gov/pubmed/15449324?tool=bestpractice.com
When other conditions with similar EEG abnormalities have been ruled out, these findings can have high specificity for prion disease.[71]Geschwind MD, Jay C. Assessment of rapidly progressive dementias. Concise review related to chapter 362: Alzheimer disease and other primary dementias. In: Braunwald E, Harrison TR, Fauci AS, et al, eds. Harrison's principles of internal medicine. New York, NY: McGraw-Hill; 2001:2391-2398.
CSF testing
The real-time quaking-induced conversion (RT-QuIC) is being used in a number of countries as a direct assay for the presence of abnormal prions in the CSF. It has very high specificity and sensitivity for CJD, particularly the sporadic forms.[68]Groveman BR, Orrú CD1, Hughson AG, et al. Extended and direct evaluation of RT-QuIC assays for Creutzfeldt-Jakob disease diagnosis. Ann Clin Transl Neurol. 2016 Dec 27;4(2):139-144.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5288466
http://www.ncbi.nlm.nih.gov/pubmed/28168213?tool=bestpractice.com
[69]Hermann P, Appleby B, Brandel JP, et al. Biomarkers and diagnostic guidelines for sporadic Creutzfeldt-Jakob disease. Lancet Neurol. 2021 Mar;20(3):235-246.
https://www.doi.org/10.1016/S1474-4422(20)30477-4
http://www.ncbi.nlm.nih.gov/pubmed/33609480?tool=bestpractice.com
[70]Centers for Disease Control and Prevention. CDC’s Diagnostic Criteria for Creutzfeldt-Jakob Disease (CJD). 2018 [internet publication].
https://www.cdc.gov/prions/cjd/diagnostic-criteria.html
The 14-3-3 protein found in CSF was previously reported as being a strong indicator of CJD; however, the sensitivity and specificity of this test vary greatly in the literature.[71]Geschwind MD, Jay C. Assessment of rapidly progressive dementias. Concise review related to chapter 362: Alzheimer disease and other primary dementias. In: Braunwald E, Harrison TR, Fauci AS, et al, eds. Harrison's principles of internal medicine. New York, NY: McGraw-Hill; 2001:2391-2398. Though there is great disagreement about the sensitivity of this test for sCJD, it is becoming more accepted in the neurology community that this test is not sufficiently specific for sCJD or other human prion diseases.[83]Chitravas N, Jung RS, Kofskey DM, et al. Treatable neurological disorders misdiagnosed as Creutzfeldt-Jakob disease. Ann Neurol. 2011 Sep;70(3):437-44.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3170496
http://www.ncbi.nlm.nih.gov/pubmed/21674591?tool=bestpractice.com
Current guidelines are shifting away from the use of this test.
Research studies have suggested that CSF proteins, such as total tau (T-tau) and neuron-specific enolase (NSE), have a slightly higher or equivalent sensitivity but much higher specificity than 14-3-3. However, these proteins can also be elevated in other rapidly progressive non-prion diseases, supporting the idea that they are released during neuronal death and injury and are not necessarily specific for prion disease.[84]Tschampa HJ, Neumann M, Zerr I, et al. Patients with Alzheimer's disease and dementia with Lewy bodies mistaken for Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 2001 Jul;71(1):33-9.
http://jnnp.bmj.com/content/71/1/33.long
http://www.ncbi.nlm.nih.gov/pubmed/11413259?tool=bestpractice.com
Although helpful in confirming rapid neuronal deterioration, these biomarkers cannot definitively diagnose or rule out prion disease.[66]Zerr I, Kallenberg K, Summers DM, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. 2009 Oct;132(Pt 10):2659-68.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2759336/?tool=pubmed
http://www.ncbi.nlm.nih.gov/pubmed/19773352?tool=bestpractice.com
[71]Geschwind MD, Jay C. Assessment of rapidly progressive dementias. Concise review related to chapter 362: Alzheimer disease and other primary dementias. In: Braunwald E, Harrison TR, Fauci AS, et al, eds. Harrison's principles of internal medicine. New York, NY: McGraw-Hill; 2001:2391-2398.[83]Chitravas N, Jung RS, Kofskey DM, et al. Treatable neurological disorders misdiagnosed as Creutzfeldt-Jakob disease. Ann Neurol. 2011 Sep;70(3):437-44.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3170496
http://www.ncbi.nlm.nih.gov/pubmed/21674591?tool=bestpractice.com
[85]Geschwind M, Haman A, Torres-Chae C, et al. CSF findings in a large United States sporadic CJD cohort. Neurology. 2007;68(suppl 1):A142.
Blood and genetic testing
No approved blood tests are available for detecting prions, but patients should be screened for genetic mutations on the PRNP gene. In the US, blood samples can be sent to the National Prion Disease Pathology Surveillance Center for analysis.
National Prion Disease Pathology Surveillance Center
Opens in new window In Canada, they can be sent to the Canadian Creutzfeldt-Jakob Disease (CJD) Surveillance System.
Public Health Agency of Canada: prion disease information
Opens in new window
It is recommended that patients and families undergo genetic counseling and understand the implications before undergoing testing and before learning genetic results. The genetic counseling protocol used for testing for Huntington's disease is typically followed for PRNP testing.[86]Huntington's Disease Society of America. Genetic testing for Huntington’s disease: its relevance and implications. 2003. [internet publication].
http://hdsa.org/wp-content/uploads/2015/03/GeneticTesting-for-HD.pdf
Biopsy
Brain biopsy is the only definitive way to diagnose sporadic prion disease antemortem. In variant CJD, a tonsil biopsy can be diagnostic.
Due to the unpredictable pattern of protein accumulation in the brain, it is possible to get a false negative from brain biopsy. The procedure may place patients at unnecessary risk for infection or further brain damage. Even if the diagnosis is confirmed, there is still no available treatment.
Prion proteins are also resistant to standard surgical sterilization methods, and medical staff performing the procedure may be placed at risk for CJD transmission. UK Department of Health guidance for minimizing transmission risk is available.[27]Department of Health. Minimise transmission risk of CJD and vCJD in healthcare settings. Nov 2021 [internet publication].
https://www.gov.uk/government/publications/guidance-from-the-acdp-tse-risk-management-subgroup-formerly-tse-working-group
Brain biopsy is only recommended when MRI is negative for CJD and all other conditions have been ruled out with other less invasive methods.[71]Geschwind MD, Jay C. Assessment of rapidly progressive dementias. Concise review related to chapter 362: Alzheimer disease and other primary dementias. In: Braunwald E, Harrison TR, Fauci AS, et al, eds. Harrison's principles of internal medicine. New York, NY: McGraw-Hill; 2001:2391-2398.
Autopsy
Autopsy is strongly encouraged, as pathologic confirmation is the only definitive way to diagnose prion disease outside of biopsy.[36]Kretzschmar HA, Ironside JW, DeArmond SJ, et al. Diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Arch Neurol. 1996 Sep;53(9):913-20.
http://www.ncbi.nlm.nih.gov/pubmed/8815857?tool=bestpractice.com