Etiology
The vast majority of cases of IgA nephropathy (IgAN) are idiopathic, but it may occur secondary to other diseases.[8]
Hepatic IgAN, in which mesangial IgA deposition is associated with chronic liver disease (particularly alcoholic cirrhosis), is the commonest form of secondary IgAN.[24][25] It is believed to be a consequence of impaired hepatic clearance of IgA. Mesangial IgA is a common autopsy finding in patients with chronic liver disease, but only a minority have clinical manifestations of kidney disease other than invisible hematuria.
IgAN has also been reported in association with HIV infection and AIDS.[26][27][28] The polyclonal increase in serum IgA, a feature of AIDS, has been cited as a predisposing factor. The strength of this association is, however, questionable; autopsy studies have indicated a prevalence of IgAN between zero and 7.75% in HIV-infected patients. IgAN is rare in IgA myeloma or other IgA monoclonal gammopathies. These observations indicate that a high circulating load of monoclonal or polyclonal IgA is not sufficient to promote mesangial IgA deposition without other abnormalities of the IgA system.
Indirect evidence suggests a close relationship between IgAN and the small vessel systemic vasculitis IgA vasculitis (previously known as Henoch-Schönlein purpura [HSP]). IgA vasculitis is characterized by small blood vessel deposition of IgA that predominantly affects the skin, joints, gut, and kidney, with nephritis that may be histologically indistinguishable from IgAN. IgAN is increasingly thought of as IgA vasculitis without the rash.[29] It is not known why some people get kidney limited disease (IgAN), while others develop systemic disease (IgA vasculitis).
Although IgAN is commonly sporadic, familial cases have been reported.[16][17][18][19] Study of the three novel loci (designated IGAN1, IGAN2, IGAN3) has identified no likely candidate genes. The 2q36 locus identified in one Canadian pedigree is potentially informative because it contains COL4A3 and COL4A4 genes coding for basement membrane collagen, mutations of which are associated with thin basement membrane nephropathy.[30] Four IgAN genome-wide association studies have shown an enrichment of single nucleotide polymorphisms (SNPs) implicated in autoimmune or inflammatory traits (multiple alleles within the HLA region at chromosome 6p21 and chromosome 1q32 suggesting a role for complement-regulatory proteins).[20][21][22][23] Furthermore, a number of loci associated with IgAN encode proteins implicated in maintenance of the intestinal barrier and regulation of mucosal immune response to pathogens.
Pathophysiology
The pathogenesis of IgA nephropathy (IgAN) is incompletely understood. It has been proposed that the initiating event is the formation of circulating IgA-containing immune complexes that are prone to mesangial deposition. In susceptible people, the deposited IgA triggers variable degrees of glomerular inflammation and subsequent glomerular and tubulointerstitial scarring.[31][32] The circulating IgA immune complexes are predominantly composed of low-affinity polymeric IgA of the IgA1 subclass. The IgA1 present in these complexes displays subtle changes in the composition of the carbohydrates attached to the IgA1 hinge region. There is a reduction in the amount of galactose present in the hinge region glycans and it has been suggested that this change triggers autoantibody production in IgAN.[33] IgA and IgG autoantibodies specific for the hinge region have been detected in the serum of IgAN cohorts from Europe and Asia and are thought to promote immune complex formation in IgAN. The role of complement activation in IgAN remains unclear, despite the frequent identification of C3 in kidney biopsies and the identification of an association between IgAN and complement-regulatory proteins in genome-wide association studies.[34][35]
Classification
A number of histopathologic classifications have been published. The most widely validated of these is the Oxford classification.[2]
The original Oxford classification, published in 2009, comprises four independent morphologic lesions that have high inter- and intra-observer reproducibility and are independently associated with a poor prognosis independent of clinical features at the time of kidney biopsy:
Mesangial hypercellularity (M0 or M1)[Figure caption and citation for the preceding image starts]: Mesangial hypercellularity in IgA nephropathy (Periodic acid-Schiff stain, x600)Courtesy of Drs Hwei Yee Lee, Cristine Ding, and Yong Howe Ho (Tan Tock Seng Hospital, Singapore) [Citation ends].

Endocapillary hypercellularity (E0 or E1)[Figure caption and citation for the preceding image starts]: Endocapillary hypercellularity in IgA nephropathy (Periodic acid–Schiff stain, x400)Courtesy of Drs Hwei Yee Lee, Cristine Ding, and Yong Howe Ho (Tan Tock Seng Hospital, Singapore) [Citation ends].
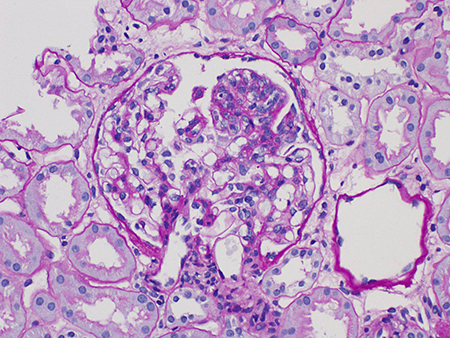
Segmental glomerulosclerosis (S0 or S1)[Figure caption and citation for the preceding image starts]: Segmental glomerulosclerosis in IgA nephropathy (Periodic acid-Schiff stain, x400)Courtesy of Drs Hwei Yee Lee, Cristine Ding, and Yong Howe Ho (Tan Tock Seng Hospital, Singapore) [Citation ends].
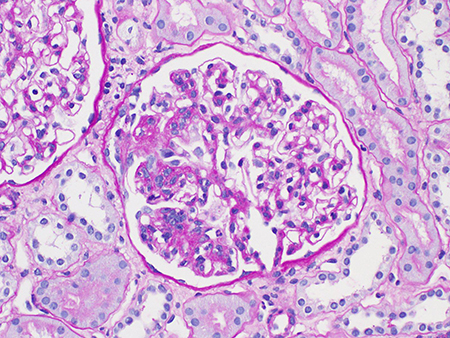
Tubular atrophy/interstitial fibrosis (T0, T1, or T2).[Figure caption and citation for the preceding image starts]: Tubular atrophy and interstitial fibrosis in IgA nephropathy (the atrophied tubules are seen on the left of the field, and normal tubules are seen on the right) (Periodic acid-Schiff stain, x400)Courtesy of Drs Hwei Yee Lee, Cristine Ding, and Yong Howe Ho (Tan Tock Seng Hospital, Singapore) [Citation ends].

Since its publication, the Oxford classification has been validated in multiple patient cohorts from North America, Europe, and Asia.[3][4][5]
In the updated Oxford Classification, published in 2017, crescents were added to the four original lesions: C0 (no crescents), C1 (crescents in less than 25% of glomeruli), and C2 (crescents in 25% or more of glomeruli), forming a new MEST-C score.[6] This is the result of a working subgroup of the IgAN Classification Working Group that demonstrated crescents (C1 or C2) were independent predictors of kidney outcomes in a pooled cohort of 3096 patients.[7] The other notable addition to the updated Oxford Classification 2017 is the subclassification of segmental sclerosis, identifying those cases with evidence of podocyte hypertrophy and/or tip lesion.
While the pathologic definition of IgAN is straightforward, the variations in phenotype are striking: these include variations in geographic incidence, clinical presentation and progression, kidney histopathology, and transplant recurrence.
There are currently no clinical classification systems for patients with IgAN that accurately reflect the varied phenotype of this complex glomerulonephritis.
At present there is no proof that the entity known as IgAN is a single "disease" (using the term "disease" in its conventional sense of a single entity sharing etiologic factors and pathogenic processes). Nor is there proof that IgAN is the same "disease" in all parts of the world.
Use of this content is subject to our disclaimer