Etiology
Accidental poisoning can occur as a result of exposure to CO from internal combustion engines (vehicle or generator), building fires, and indoor sources such as poorly vented stoves or heaters. CO poisoning is more common during the winter months when heating systems are in use and windows are kept closed. There are increased cases of CO poisoning after natural disasters, often associated with the loss of electricity and the improper placement of portable electric generators.[13] Other sources of CO exposure include combustion of carbonaceous fuels (e.g., gasoline, natural gas, kerosene, oil, boat exhaust, gas-powered stoves in outdoor areas, pickup truck exhaust), paint removers, and aerosol propellants. Intentional CO poisoning may be due to exposure to automobile exhaust in an enclosed garage, with the CO being formed from incomplete combustion of carbon compounds.[6][12][14][15][16][17]
[Figure caption and citation for the preceding image starts]: Common sources of carbon monoxideAshcroft J, et al. Carbon monoxide poisoning. BMJ 2019;365:I2299 (used with permission) [Citation ends].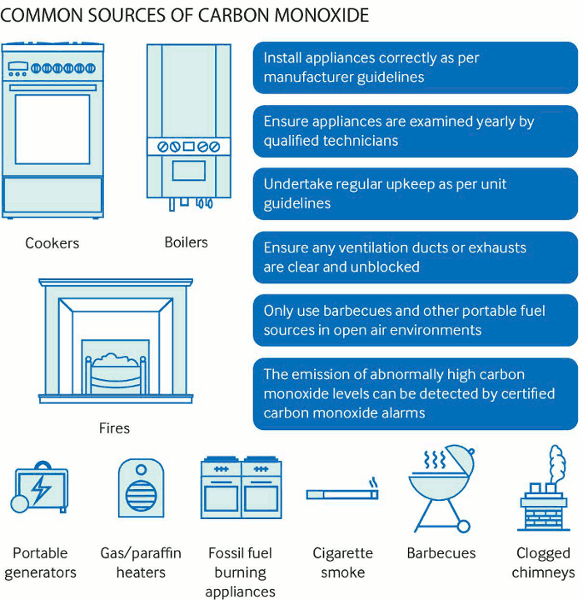
Pathophysiology
The clinical manifestations of CO poisoning mostly result from the interaction of the CO molecule with heme-containing proteins, and the ensuing cellular effects.
Hemoglobin-related effects
CO binds to hemoglobin with 250-fold greater affinity than oxygen.[4] This preferential binding of CO to hemoglobin displaces oxygen from the hemoglobin molecule and leads to the formation of carboxyhemoglobin (CO-Hb).[4] This displaces oxygen from the hemoglobin molecule. CO binding to one molecule of hemoglobin stabilizes the relaxed state of the hemoglobin tetramer. This increases oxygen affinity of the other hemoglobin molecules, which shifts the oxyhemoglobin dissociation curve "to the left", and thereby decreases the amount of oxygen available to cells at a given oxygen pressure. These effects combine to reduce oxygen delivery.[2][4][18]
Cytochrome c oxidase effects
CO also binds to cytochrome c oxidase in the electron transport chain. This disrupts oxidative phosphorylation, halting the transfer of electrons within the electron transport chain. Excess electrons upstream can then lead to superoxide formation and cellular damage. Additionally, the binding of CO to the second heme center of cytochrome c oxidase stops the creation of the proton gradient that feeds adenosine triphosphate (ATP) synthase, which limits intracellular energy transfer by lowering the availability of ATP. The ensuing cascade furthers exacerbates cellular damage.[4][19][20][21]
Studies also report biochemical and antigenic changes in major basic protein. This, in combination with products of lipid peroxidation, may lead to immunologic cascade activation. This results in cell damage. Other suggested mechanisms of cell injury and death include glutamate-mediated neuronal injury, atherogenesis, cytochrome P-450 involvement, and apoptosis.[16]
Use of this content is subject to our disclaimer