Approach
MG most commonly affects young adult women (under 40 years) and older men (over 60 years), but it can occur at any age.[36][37]
Following a review of the patient's medical history and typical exam findings, the diagnosis of MG can be established by clinical and serologic testing. If serologic testing is unremarkable, clinical neurophysiologic testing should be performed. Pulmonary function tests can help predict whether respiration is impaired, and so help avoid myasthenic crisis.
History
Typical features are weakness and fatigability of skeletal muscles with a characteristic distribution. The disease usually presents with one of three different forms: ocular, oropharyngeal, or generalized.
Depending on the predominant initial form, patients with MG may have a multitude of symptoms including ptosis, diplopia, dysarthria (speech disorder), dysphagia (difficulty swallowing), facial paresis, proximal limb weakness, and shortness of breath. Characteristically the limb weakness worsens with activity (fatigue) and improves on rest, and the fluctuations often (but not always) show diurnal variation (better in the morning than in the evening).
Less commonly, patients may present with predominant or exclusive weakness of the limbs, sometimes called "limb-girdle" MG.
Physical exam
The clinical severity of MG is usually graded functionally and regionally.[4][68][69]
Fatigable muscle weakness may be localized or generalized involving the eyelids, extraocular eye muscles, face, oropharynx, neck, respiratory muscles, and extremities. Ptosis and diplopia occur early in the majority of patients.[22] Ptosis time (which is normally more than 3 minutes in healthy people) is checked by asking the patient to look up and recording the time until ptosis develops. Pulling up on the upper eyelid may induce ptosis in the contralateral lid. Cooling of the eyelid for 2 to 5 minutes with an ice pack improves ptosis in >95% of patients with MG, but may not improve severe ptosis.[70][71]
Between 50% and 60% of patients who present with purely ocular symptoms will progress to develop generalized MG, and the vast majority will do so within the first 1 to 2 years.[23][Figure caption and citation for the preceding image starts]: Ptosis of the left upper eyelid in a patient with myasthenia gravisForoozan R, Sambursky R. Ocular myasthenia gravis and inflammatory bowel disease: a case report and literature review. Br J Ophthalmol. 2003 Sep;87(9):1186-7. [Citation ends].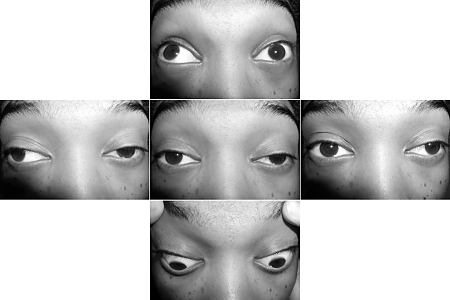
When the facial and oropharyngeal muscles are affected, there may be a characteristic flattened or transverse smile, or nasal speech, and difficulty in chewing and swallowing.
Examination of the limbs shows proximal muscle weakness with fatigue. Arm abduction time is checked by asking patients to hold their arms outstretched; healthy people can typically hold their arms up for more than 3 minutes.
There should be no evident muscle wasting (with the exception of the tongue and less commonly the palate in MG patients with antibodies to muscle-specific tyrosine kinase [MuSK]), and reflexes are normal. Sensations are intact (unless another disorder is also present), and there is no autonomic dysfunction.
In cases of myasthenic crisis there is severe respiratory involvement and/or bulbar involvement.
Features of MuSK-MG
Patients with MuSK-MG present with one of three phenotypes; the first two are considered characteristic:[5][24]
Severe faciopharyngeal weakness, with atrophy of involved muscles in longstanding disease
Predominant neck and respiratory weakness with frequent progression to myasthenic crisis, or
Clinical features indistinguishable from non-MuSK-MG.
The following clinical features distinguish MuSK-MG from non-MuSK-MG:[5]
Ocular features are not common but can occur.
Facial and tongue atrophy is present by clinical exam and by magnetic resonance imaging (MRI).
Limb weakness is mild.
Patients with prominent neck and respiratory muscle weakness may have normal electrodiagnostic testing in face and limb muscles. Testing of involved muscles is required to establish the diagnosis.
Weakness tends to be more severe; 23% of patients progressed to crisis and 85% had a maximum Myasthenia Gravis Foundation of America (MGFA) class of III or greater in one clinic.
Poor clinical response to cholinesterase inhibitor therapy.
Features of LRP4, cortactin, agrin, and collagen Q MG
Patients with MG and antibodies to LRP4 are reported to usually have mild to moderate MG, similar to some patients with acetylcholine receptor (AChR) antibodies; the full spectrum of symptom severity was reported in one study.[9][10][13][50][72] Additional studies are needed to fully characterize clinical phenotypes, as well as any patterns of age of onset, sex differences, and ethnic/racial groups, for LRP4.
A small number of patients with antibodies to cortactin and no detectable antibodies to AChR (using standard testing) or MuSK have been found to have mild to moderate disease, including ocular MG, similar to the pattern seen in AChR-MG.[17][18][19][21] It is unknown if any of these patients had antibodies to LRP4, agrin, or collagen Q, or antibodies to AChR that would be detected only with cell-based clustered AChR technology.
The features of MG in patients with antibodies to agrin or collagen Q have yet to be established.
Overview of investigations
MG is a clinical diagnosis supported by serologic, electrophysiologic, and pharmacologic tests.
Serologic testing
All patients should have serologic testing.
AChR antibody standard binding and modulating assay: antibodies to AChR are detected in 80% to 90% of patients with generalized MG and up to 50% of patients with ocular MG, with 99% specificity in both cases.[70] Absolute titer does not correlate with the severity of MG, although high levels of modulatory AChR antibodies have been suggested to correlate with severity in some studies.[2][22]
AChR antibody cell-based assay: identifies patients with MG who in general appear similar to patients identified by standard AChR testing, with an increased percentage of prepubertal onset disease.[73]
MuSK antibody assay: if the standard test for AChR antibodies is negative, proceed with MuSK antibody serologic testing. MuSK antibodies are detected in up to 70% of patients with generalized MG who are seronegative for AChR antibodies.[5][62] In the US, MuSK antibodies are common in the black population; 70% of black women with generalized seronegative MG have MuSK antibodies.[5]
Striational receptor antibody assays: assays for antibodies to titin and ryanodine are not done routinely in all patients, and the antibodies are rare in patients without thymoma, other than "elderly onset" patients.[74] They are detected in 75% to 95% of patients with thymoma and MG, and their presence may indicate recurrence of thymoma.[75]
Testing for antibodies to LRP4, agrin, collagen Q, or cortactin is not widely available, and it is not clear how specific these antibodies are for MG, but LRP4 antibodies may prove useful in the appropriate clinical setting.[10] Antibodies to cortactin are seen in myopathies, suggesting that testing for cortactin antibodies would not be useful as a diagnostic test for MG.[17][19]
Electrophysiologic testing
If serologic testing is unremarkable or there are atypical clinical features, electrophysiologic testing should be performed.
Repetitive nerve stimulation (RNS) at a slow rate should reveal a decrement. In this procedure, electric shocks are delivered to the nerve, and action potentials are recorded from surface electrodes over the muscle.[70] Improvement of a baseline decrement after brief exercise (post-exercise facilitation) and enhanced decrement at 2 to 3 minutes post-exercise (post-exercise exhaustion) are typical electrodiagnostic features. Sensitivity is 79% in generalized MG and 50% in ocular MG, with a specificity of 97%.[70] A decrement of 7% to 8% has been suggested as a cutoff for facial muscles.[76] Increasing the number of muscles tested increases the chance of demonstrating significant decrement.[77]
If RNS is negative or equivocal, single-fiber EMG (SFEMG) is recommended. SFEMG is a measure of neuromuscular junction (NMJ) transmission between two or more adjacent muscle fibers innervated by the same motor axon. SFEMG may show an increased variability in motor latencies (jitter) or complete failure of NMJ transmission (block) in muscle fibers. When performed on facial muscles, the test is positive in 86% to 92% of patients with ocular MG, with a specificity of 70% to 96%. In patients with generalized MG, specificity and sensitivity are reported to be as high as 98%.[70] This test is not specific for MG, and it is important to be certain that there is no evidence of denervation or myopathy in the tested muscles.
Imaging
Computed tomography (CT) scan of the chest should be performed for all newly diagnosed patients to detect thymoma (which occurs in about 15% of patients with MG) or thymic hyperplasia (which occurs in 75% of MG patients). [Figure caption and citation for the preceding image starts]: Computed tomography showing anterior mediastinal massHamid UI, Jones JM. Mediastinal mass. BMJ Case Reports 2010 Oct 4;2010:bcr1120092471. [Citation ends].
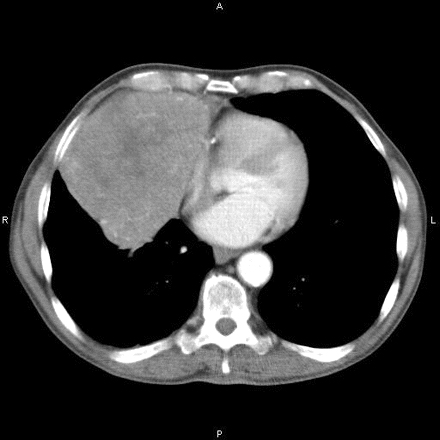
Routine MRI offers no added benefit. Chemical-shift and diffusion-weighted MRI may be able to identify germinal center follicular hyperplasia and distinguish this from normal thymus.[78][79]
Other tests to consider
The "ice pack test" can be a useful diagnostic test for a patient with significant ptosis. Cooling of the eyelid for 2 to 5 minutes with an ice pack improves ptosis in >95% of patients with MG, but may not improve severe ptosis.[70][71]
Edrophonium (tensilon) test is largely of historical interest but may be used occasionally by neuro-ophthalmologists and neurologists for evaluation of ophthalmoparesis and/or isolated ptosis.
Patients with certain forms of congenital MG may not always present in infancy or early childhood. They will usually be seronegative (there are rare reports of MuSK antibody in a few families) but may have abnormal neurophysiologic testing, sometimes different from, but in some types identical to, that for autoimmune MG. If there are concerns, genetic testing for congenital myasthenic syndromes can be performed.
Myasthenic crisis
Myasthenic crisis is a medical emergency. It is defined as an MG exacerbation requiring mechanical ventilation. Indications for mechanical ventilation are forced vital capacity (FVC) of 15 mL/kg or less (normal ≥60 mL/kg) and/or negative inspiratory force (NIF) of 20 cm H₂O or less (normal ≥70 cm H₂O). Serial measurements of FVC and NIF are taken.
A high index of clinical suspicion is necessary for myasthenic crisis because patients may initially appear stable.[23][80] Physicians should not wait for abnormal arterial blood gas (ABG) as it occurs late in the course after clinical decompensation. Clinical judgment should always supplement, and if need be supersede, respiratory parameters.[81]
Myasthenic crisis may be provoked by infections (particularly respiratory infections), aspiration, medications including high-dose corticosteroids, surgery, medications that are contraindicated or relatively contraindicated in MG, failure to adhere to medications, administration of immune checkpoint inhibitors as cancer therapy, or trauma.[57][58][80][81][82] Older patients and patients who also have other autoimmune diseases appear to be at increased risk of developing myasthenic crisis.[83]
Appropriate measures should be taken to eliminate provoking factors. Oropharyngeal muscle weakness may also contribute to respiratory failure.
How to take a venous blood sample from the antecubital fossa using a vacuum needle.
Use of this content is subject to our disclaimer