Cushing disease (pituitary adenoma)
Cushing disease is caused by adrenocorticotropic hormone (ACTH)-secreting pituitary tumors. The ultimate goal of therapy is to remove the causative pituitary adenoma and normalize cortisol levels while preserving pituitary function.
First-line therapy is transsphenoidal (TSS) resection of the causative pituitary adenoma performed by an experienced surgeon.[26]Fleseriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing's disease: a guideline update. Lancet Diabetes Endocrinol. 2021 Dec;9(12):847-75.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8743006
http://www.ncbi.nlm.nih.gov/pubmed/34687601?tool=bestpractice.com
[69]Nieman LK, Biller BM, Findling JW, et al. Treatment of Cushing’s syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2015 Aug;100(8):2807-31.
https://academic.oup.com/jcem/article/100/8/2807/2836065
http://www.ncbi.nlm.nih.gov/pubmed/26222757?tool=bestpractice.com
[72]Pivonello R, De Leo M, Cozzolino A, et al. The treatment of Cushing's disease. Endocr Rev. 2015 Aug;36(4):385-486.
https://academic.oup.com/edrv/article/36/4/385/2354703
http://www.ncbi.nlm.nih.gov/pubmed/26067718?tool=bestpractice.com
Surgery can be done using an endoscopic or microscopic approach. Results are comparable between both techniques for microadenomas.[73]Broersen LHA, Biermasz NR, van Furth WR, et al. Endoscopic vs. microscopic transsphenoidal surgery for Cushing's disease: a systematic review and meta-analysis. Pituitary. 2018 Oct;21(5):524-34.
https://link.springer.com/article/10.1007%2Fs11102-018-0893-3
http://www.ncbi.nlm.nih.gov/pubmed/29767319?tool=bestpractice.com
Whether there is potential incremental benefit with an endoscopic approach for macroadenomas remains unclear.[26]Fleseriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing's disease: a guideline update. Lancet Diabetes Endocrinol. 2021 Dec;9(12):847-75.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8743006
http://www.ncbi.nlm.nih.gov/pubmed/34687601?tool=bestpractice.com
[73]Broersen LHA, Biermasz NR, van Furth WR, et al. Endoscopic vs. microscopic transsphenoidal surgery for Cushing's disease: a systematic review and meta-analysis. Pituitary. 2018 Oct;21(5):524-34.
https://link.springer.com/article/10.1007%2Fs11102-018-0893-3
http://www.ncbi.nlm.nih.gov/pubmed/29767319?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Algorithm for the treatment of Cushing disease (DST = dexamethasone suppression test. IPSS = inferior petrosal sinus sampling. ACTH = adrenocorticotropic hormone. *Pituitary surgery should be performed by an experienced surgeon. †Absence of ACTH-staining adenoma. §Lifelong monitoring for hypopituitarism and secondary neoplasia in the radiation field required. ¶On maximum tolerated dose of the drug)Fleseriu M et al. Lancet Diabetes Endocrinol. 2021 Dec;9(12):847-75; used with permission [Citation ends].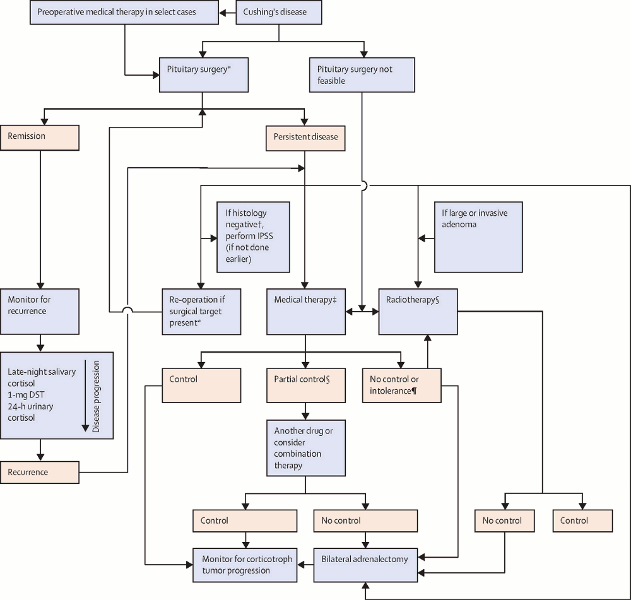
Many patients are supported with corticosteroids following pituitary surgery but should be assessed for remission during the first postoperative week.[71]Hammer GD, Tyrrell JB, Lamborn KR, et al. Transsphenoidal microsurgery for Cushing's disease: initial outcome and long-term results. J Clin Endocrinol Metab. 2004 Dec;89(12):6348-57.
https://academic.oup.com/jcem/article/89/12/6348/2844760
http://www.ncbi.nlm.nih.gov/pubmed/15579802?tool=bestpractice.com
This is done by measuring morning cortisol at least 24 hours after the last dose of corticosteroid therapy. Patients with a postoperative morning cortisol of <2 micrograms/dL are considered to be in remission and can transition into long-term follow-up. Patients with a postoperative morning cortisol of >5 micrograms/dL require further evaluation and possibly further therapy. Patients with a morning cortisol between 2 and 5 micrograms/dL should be followed with additional measurements to detect a drop in subsequent morning cortisol levels. Individuals with morning cortisols >2 micrograms/dL after surgery are 2.5 times more likely to have recurrences than those with cortisol levels <2 micrograms/dL.[74]Abu Dabrh AM, Singh Ospina NM, Al Nofal A, et al. Predictors of biochemical remission and recurrence after surgical and radiation treatments of Cushing disease: a systematic disease: a systematic review and meta-analysis. Endocr Pract. 2016 Apr;22(4):466-75.
http://www.ncbi.nlm.nih.gov/pubmed/26789343?tool=bestpractice.com
[75]Patil CG, Prevedello DM, Lad SP, et al. Late recurrences of Cushing's disease after initial successful transsphenoidal surgery. J Clin Endocrinol Metab. 2008 Feb;93(2):358-62.
https://academic.oup.com/jcem/article/93/2/358/2597991
http://www.ncbi.nlm.nih.gov/pubmed/18056770?tool=bestpractice.com
[76]Fleseriu M, Hamrahian AH, Hoffman AR, et al; AACE Neuroendocrine and Pituitary Scientific Committee. American Association of Clinical Endocrinologists and American College of Endocrinology Disease State Clinical Review: diagnosis of recurrence in Cushing disease. Endocr Pract. 2016 Dec;22(12):1436-48.
http://www.ncbi.nlm.nih.gov/pubmed/27643842?tool=bestpractice.com
Postoperative hypocortisolism is predictive of remission, hence some centers advocate withholding routine corticosteroid therapy after pituitary surgery and monitoring cortisol levels every 8 hours or if symptoms of adrenal insufficiency occur.[77]Sughrue ME, Shah JK, Devin JK, et al. Utility of the immediate postoperative cortisol concentrations in patients with Cushing's disease. Neurosurgery. 2010 Sep;67(3):688-95.
http://www.ncbi.nlm.nih.gov/pubmed/20651632?tool=bestpractice.com
If adrenal insufficiency occurs or low cortisol levels are documented, corticosteroid therapy is initiated. Other centers begin routine corticosteroid therapy immediately after surgery and evaluate for remission of hypercortisolism later in the postoperative course.[78]Hameed N, Yedinak CG, Brzana J, et al. Remission rate after transsphenoidal surgery in patients with pathologically confirmed Cushing's disease, the role of cortisol, ACTH assessment and immediate reoperation: a large single center experience. Pituitary. 2013 Dec;16(4):452-8.
http://www.ncbi.nlm.nih.gov/pubmed/23242860?tool=bestpractice.com
Corticosteroids are usually rapidly tapered to physiologic doses within 1 week or less (often by discharge from the hospital). Testing to see if the hypothalamic-pituitary-adrenal (HPA) axis has recovered can be done in follow-up by 3 months after surgery. Testing is usually a morning cortisol prior to the patient taking the morning hydrocortisone dose, if hydrocortisone therapy had been continued. Cortisol levels of >20 micrograms/dL indicate recovery of the axis. Levels <3 micrograms/dL indicate a continued need for corticosteroids. Levels between 3 micrograms/dL and 20 micrograms/dL should prompt further testing (cosyntropin stimulation testing, insulin tolerance testing, or metyrapone testing). Once recovery of HPA axis has been established, patients need to undergo testing for possible recurrence. Salivary cortisol testing seems to be a better predictor of early recurrence.[76]Fleseriu M, Hamrahian AH, Hoffman AR, et al; AACE Neuroendocrine and Pituitary Scientific Committee. American Association of Clinical Endocrinologists and American College of Endocrinology Disease State Clinical Review: diagnosis of recurrence in Cushing disease. Endocr Pract. 2016 Dec;22(12):1436-48.
http://www.ncbi.nlm.nih.gov/pubmed/27643842?tool=bestpractice.com
[79]Danet-Lamasou M, Asselineau J, Perez P, et al. Accuracy of repeated measurements of late-night salivary cortisol to screen for early-stage recurrence of Cushing's disease following pituitary surgery. Clin Endocrinol (Oxf). 2015 Feb;82(2):260-6.
http://www.ncbi.nlm.nih.gov/pubmed/24975391?tool=bestpractice.com
Additional therapy should be considered in patients with failure of initial pituitary surgery or with recurrence of disease. The incidence of recurrence in Cushing disease is high, with 50% of recurrences occurring during the first 50 months after first surgery.[80]Braun LT, Rubinstein G, Zopp S, et al. Recurrence after pituitary surgery in adult Cushing's disease: a systematic review on diagnosis and treatment. Endocrine. 2020 Nov;70(2):218-31.
https://www.doi.org/10.1007/s12020-020-02432-z
http://www.ncbi.nlm.nih.gov/pubmed/32743767?tool=bestpractice.com
Standard therapies include repeat pituitary surgery, radiation therapy, bilateral adrenalectomy, or medical therapy.[72]Pivonello R, De Leo M, Cozzolino A, et al. The treatment of Cushing's disease. Endocr Rev. 2015 Aug;36(4):385-486.
https://academic.oup.com/edrv/article/36/4/385/2354703
http://www.ncbi.nlm.nih.gov/pubmed/26067718?tool=bestpractice.com
[81]Reincke M, Ritzel K, Osswald A, et al. A critical re-appraisal of bilateral adrenalectomy for ACTH-dependent Cushing's syndrome. Eur J Endocrinol. 2015 Oct;173(4):M23-32.
https://eje.bioscientifica.com/view/journals/eje/173/4/M23.xml
http://www.ncbi.nlm.nih.gov/pubmed/25994948?tool=bestpractice.com
Success rates of these treatment options vary between 25% (for some of the medical therapies) and 100% (bilateral adrenalectomy). Treatment options have specific advantages, limitations, and side-effects so treatment decisions should be individualized according to the specific needs of the patient and risk of complications.[80]Braun LT, Rubinstein G, Zopp S, et al. Recurrence after pituitary surgery in adult Cushing's disease: a systematic review on diagnosis and treatment. Endocrine. 2020 Nov;70(2):218-31.
https://www.doi.org/10.1007/s12020-020-02432-z
http://www.ncbi.nlm.nih.gov/pubmed/32743767?tool=bestpractice.com
[82]Cuevas-Ramos D, Fleseriu M. Treatment of Cushing's disease: a mechanistic update. J Endocrinol. 2014 Nov;223(2):R19-39.
https://joe.bioscientifica.com/view/journals/joe/223/2/R19.xml
http://www.ncbi.nlm.nih.gov/pubmed/25134660?tool=bestpractice.com
Re-operation is frequently the preferred therapy if initial surgery fails. It should be considered in all patients with recurrence or persistence of disease. It is effective in about two-thirds of patients.[83]Friedman RB, Oldfield EH, Nieman LK, et al. Repeat transsphenoidal surgery for Cushing's disease. J Neurosurg. 1989 Oct;71(4):520-7.
http://www.ncbi.nlm.nih.gov/pubmed/2552045?tool=bestpractice.com
[84]Benveniste RJ, King WA, Walsh J, et al. Repeated transsphenoidal surgery to treat recurrent or residual pituitary adenoma. J Neurosurg. 2005 Jun;102(6):1004-12.
http://www.ncbi.nlm.nih.gov/pubmed/16028758?tool=bestpractice.com
[85]Hofmann BM, Hlavac M, Kreutzer J, et al. Surgical treatment of recurrent Cushing's disease. Neurosurgery. 2006 Jun;58(6):1108-18.
http://www.ncbi.nlm.nih.gov/pubmed/16723890?tool=bestpractice.com
However, the risk of pituitary deficiency after re-operation is 50%. This is significantly higher than after initial surgical therapy.[83]Friedman RB, Oldfield EH, Nieman LK, et al. Repeat transsphenoidal surgery for Cushing's disease. J Neurosurg. 1989 Oct;71(4):520-7.
http://www.ncbi.nlm.nih.gov/pubmed/2552045?tool=bestpractice.com
If re-operation is ineffective, or if a patient is not a candidate for re-operation, another modality should be considered.
Radiation therapy by conventional fractionated radiation therapy or stereotactic radiosurgery is most commonly used in patients with persistent hypercortisolism after incomplete corticotroph tumor resection, particularly if the tumor is aggressive or invasive or considered unresectable.[26]Fleseriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing's disease: a guideline update. Lancet Diabetes Endocrinol. 2021 Dec;9(12):847-75.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8743006
http://www.ncbi.nlm.nih.gov/pubmed/34687601?tool=bestpractice.com
Radiation therapy allows for control of hypercortisolism within 3 to 5 years in over half of patients.[86]Sonino N, Zielezny M, Fava GA, et al. Risk factors and long-term outcome in pituitary-dependent Cushing's disease. J Clin Endocrinol Metab. 1996 Jul;81(7):2647-52.
https://academic.oup.com/jcem/article/81/7/2647/2875568
http://www.ncbi.nlm.nih.gov/pubmed/8675592?tool=bestpractice.com
[87]Castinetti F, Nagai M, Dufour H, et al. Gamma knife radiosurgery is a successful adjunctive treatment in Cushing's disease. Eur J Endocrinol. 2007 Jan;156(1):91-8.
https://eje.bioscientifica.com/view/journals/eje/156/1/1560091.xml
http://www.ncbi.nlm.nih.gov/pubmed/17218730?tool=bestpractice.com
[88]Devin JK, Allen GS, Cmelak AJ, et al. The efficacy of linear accelerator radiosurgery in the management of patients with Cushing's disease. Stereotact Funct Neurosurg. 2004;82(5-6):254-62.
http://www.ncbi.nlm.nih.gov/pubmed/15665560?tool=bestpractice.com
[89]Estrada J, Boronat M, Mielgo M, et al. The long-term outcome of pituitary irradiation after unsuccessful transsphenoidal surgery in Cushing's disease. N Engl J Med. 1997 Jan 16;336(3):172-7.
https://www.nejm.org/doi/full/10.1056/NEJM199701163360303
http://www.ncbi.nlm.nih.gov/pubmed/8988897?tool=bestpractice.com
[90]Starke RM, Williams BJ, Vance ML, et al. Radiation therapy and stereotactic radiosurgery for the treatment of Cushing's disease: an evidence-based review. Curr Opin Endocrinol Diabetes Obes. 2010 Aug;17(4):356-64.
http://www.ncbi.nlm.nih.gov/pubmed/20531182?tool=bestpractice.com
This modality is perhaps best used as part of the therapy for patients with mild residual hypercortisolism, as full effects of therapy can take several years to be realized. Radiation of tumors located close to the optic chiasm increases the risk of damage to the optic chiasm, and this risk should be considered prior to therapy. Hypopituitarism can also occur following radiation therapy and is another risk to consider prior to initiating therapy. The exact risk of hypopituitarism after conventional fractionated radiation therapy versus stereotactic radiosurgery is unclear, but they seem similar.
Patients with unsuccessful re-operation who manifest severe hypercortisolism should be evaluated for therapy with medical therapy, bilateral adrenalectomy, or a combination of these therapies. The advantage to these approaches is the rapid onset of decreased cortisol levels or blockade of cortisol action.
Medical treatment with a somatostatin analog (pasireotide), a dopamine agonist (cabergoline), a steroidogenesis inhibitor (osilodrostat, ketoconazole, levoketoconazole, metyrapone, mitotane, and etomidate), or a glucocorticoid receptor antagonist (mifepristone) has been increasingly used in clinical practice. Medical therapy is indicated to control cortisol secretion in patients with mild hypercortisolism, as a short term adjunct for severe hypercortisolism before other therapies are undertaken, preoperatively to bridge the time period until control of hypercortisolism is achieved by radiation therapy, in cases with persistent or recurrent hypercortisolism after surgery, or where surgery is declined or not feasible (e.g., high surgical risk, metastatic disease).[91]Varlamov EV, McCartney S, Fleseriu M. Functioning pituitary adenomas - current treatment options and emerging medical therapies. Eur Endocrinol. 2019 Apr;15(1):30-40.
https://www.touchendocrinology.com/pituitary/journal-articles/functioning-pituitary-adenomas-current-treatment-options-and-emerging-medical-therapies
http://www.ncbi.nlm.nih.gov/pubmed/31244908?tool=bestpractice.com
[92]Feelders RA, Newell-Price J, Pivonello R, et al. Advances in the medical treatment of Cushing's syndrome. Lancet Diabetes Endocrinol. 2019 Apr;7(4):300-12.
http://www.ncbi.nlm.nih.gov/pubmed/30033041?tool=bestpractice.com
[93]Hinojosa-Amaya JM, Cuevas-Ramos D, Fleseriu M. Medical management of Cushing's syndrome: current and emerging treatments. Drugs. 2019 Jun;79(9):935-56.
http://www.ncbi.nlm.nih.gov/pubmed/31098899?tool=bestpractice.com
[94]Broersen LHA, Jha M, Biermasz NR, et al. Effectiveness of medical treatment for Cushing's syndrome: a systematic review and meta-analysis. Pituitary. 2018 Dec;21(6):631-41.
https://link.springer.com/article/10.1007%2Fs11102-018-0897-z
http://www.ncbi.nlm.nih.gov/pubmed/29855779?tool=bestpractice.com
However, there is a paucity of high-quality studies of medical therapy in Cushing disease, and caution should be employed when comparing efficacy rates owing to the variability in study design and quality.[94]Broersen LHA, Jha M, Biermasz NR, et al. Effectiveness of medical treatment for Cushing's syndrome: a systematic review and meta-analysis. Pituitary. 2018 Dec;21(6):631-41.
https://link.springer.com/article/10.1007%2Fs11102-018-0897-z
http://www.ncbi.nlm.nih.gov/pubmed/29855779?tool=bestpractice.com
Individualize medical therapy for patients with Cushing disease based on the clinical scenario, including the severity of hypercortisolism.[26]Fleseriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing's disease: a guideline update. Lancet Diabetes Endocrinol. 2021 Dec;9(12):847-75.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8743006
http://www.ncbi.nlm.nih.gov/pubmed/34687601?tool=bestpractice.com
In patients with severe disease, treat aggressively to normalize cortisol concentrations (or cortisol action). Use multiple serial tests of both urinary free cortisol and late-night salivary cortisol to monitor treatment outcomes.
Pasireotide is a somatostatin analog that selectively targets somatostatin receptors in corticotroph adenomas and is now being used to medically treat Cushing disease.[92]Feelders RA, Newell-Price J, Pivonello R, et al. Advances in the medical treatment of Cushing's syndrome. Lancet Diabetes Endocrinol. 2019 Apr;7(4):300-12.
http://www.ncbi.nlm.nih.gov/pubmed/30033041?tool=bestpractice.com
[93]Hinojosa-Amaya JM, Cuevas-Ramos D, Fleseriu M. Medical management of Cushing's syndrome: current and emerging treatments. Drugs. 2019 Jun;79(9):935-56.
http://www.ncbi.nlm.nih.gov/pubmed/31098899?tool=bestpractice.com
Pasireotide binds to a wide variety of somatostatin receptors, but with greater affinity to receptor 5, which is predominantly expressed in patients with corticotroph adenomas. The use of the regular-release formulation of pasireotide has been shown to decrease cortisol levels in most patients with Cushing disease, but only normalizes cortisol levels in 25% of patients.[95]Colao A, Petersenn S, Newell-Price J, et al. A 12-month phase 3 study of pasireotide in Cushing's disease. N Engl J Med. 2012 Mar 8;366(10):914-24.
https://www.nejm.org/doi/full/10.1056/NEJMoa1105743
http://www.ncbi.nlm.nih.gov/pubmed/22397653?tool=bestpractice.com
The long-acting formulation of pasireotide showed normalization in approximately 40% of patients, but hyperglycemia was noted in up to 80% of patients.[96]Lacroix A, Gu F, Gallardo W, et al. Efficacy and safety of once-monthly pasireotide in Cushing's disease: a 12 month clinical trial. Lancet Diabetes Endocrinol. 2018 Jan;6(1):17-26.
http://www.ncbi.nlm.nih.gov/pubmed/29032078?tool=bestpractice.com
In one study, tumor shrinkage was noted in 62.5% of patients after 6 months of pasireotide treatment.[97]Simeoli C, Auriemma RS, Tortora F, et al. The treatment with pasireotide in Cushing's disease: effects of long-term treatment on tumor mass in the experience of a single center. Endocrine. 2015 Dec;50(3):725-40.
http://www.ncbi.nlm.nih.gov/pubmed/25743263?tool=bestpractice.com
Salivary cortisol also decreased after treatment; thus, salivary cortisol may be a more convenient biomarker to follow in assessing response to treatment in patients with Cushing disease.[98]Findling JW, Fleseriu M, Newell-Price J, et al. Late-night salivary cortisol may be valuable for assessing treatment response in patients with Cushing's disease: 12-month, phase III pasireotide study. Endocrine. 2016 Nov;54(2):516-23.
https://link.springer.com/article/10.1007%2Fs12020-016-0978-6
http://www.ncbi.nlm.nih.gov/pubmed/27209465?tool=bestpractice.com
One 10-year single-center experience with pasireotide reported sustained biochemical and clinical benefit in about 60% of patients with Cushing disease.[99]Trementino L, Michetti G, Angeletti A, et al. A single-center 10-year experience with pasireotide in Cushing's disease: patients' characteristics and outcome. Horm Metab Res. 2016 May;48(5):290-8.
https://www.thieme-connect.com/products/ejournals/html/10.1055/s-0042-101347
http://www.ncbi.nlm.nih.gov/pubmed/27127913?tool=bestpractice.com
This is a pituitary-targeted therapy and should only be used in patients with hypercortisolism due to ACTH-secreting pituitary tumors.[100]Petersenn S, Fleseriu M. Pituitary-directed medical therapy in Cushing's disease. Pituitary. 2015 Apr;18(2):238-44.
http://www.ncbi.nlm.nih.gov/pubmed/25627118?tool=bestpractice.com
There is a high risk of hyperglycemia, which requires careful patient selection, and a risk of QTc prolongation.[26]Fleseriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing's disease: a guideline update. Lancet Diabetes Endocrinol. 2021 Dec;9(12):847-75.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8743006
http://www.ncbi.nlm.nih.gov/pubmed/34687601?tool=bestpractice.com
Cabergoline, a dopamine agonist, has been used off-label in the treatment of Cushing disease in some countries with limited results.[92]Feelders RA, Newell-Price J, Pivonello R, et al. Advances in the medical treatment of Cushing's syndrome. Lancet Diabetes Endocrinol. 2019 Apr;7(4):300-12.
http://www.ncbi.nlm.nih.gov/pubmed/30033041?tool=bestpractice.com
[93]Hinojosa-Amaya JM, Cuevas-Ramos D, Fleseriu M. Medical management of Cushing's syndrome: current and emerging treatments. Drugs. 2019 Jun;79(9):935-56.
http://www.ncbi.nlm.nih.gov/pubmed/31098899?tool=bestpractice.com
[101]Palui R, Sahoo J, Kamalanathan S, et al. Effect of cabergoline monotherapy in Cushing's disease: an individual participant data meta-analysis. J Endocrinol Invest. 2018 Dec;41(12):1445-55.
http://www.ncbi.nlm.nih.gov/pubmed/30097903?tool=bestpractice.com
Steroidogenesis inhibitors (agents that decrease adrenal corticosteroid production, such as osilodrostat, ketoconazole, levoketoconazole, metyrapone, mitotane, and etomidate) can be used (though off-label in some countries, except osilodrostat) in the treatment of Cushing syndrome.[26]Fleseriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing's disease: a guideline update. Lancet Diabetes Endocrinol. 2021 Dec;9(12):847-75.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8743006
http://www.ncbi.nlm.nih.gov/pubmed/34687601?tool=bestpractice.com
[92]Feelders RA, Newell-Price J, Pivonello R, et al. Advances in the medical treatment of Cushing's syndrome. Lancet Diabetes Endocrinol. 2019 Apr;7(4):300-12.
http://www.ncbi.nlm.nih.gov/pubmed/30033041?tool=bestpractice.com
[93]Hinojosa-Amaya JM, Cuevas-Ramos D, Fleseriu M. Medical management of Cushing's syndrome: current and emerging treatments. Drugs. 2019 Jun;79(9):935-56.
http://www.ncbi.nlm.nih.gov/pubmed/31098899?tool=bestpractice.com
Osilodrostat is a potent oral inhibitor of steroidogenesis (inhibits steroid 11-beta-hydroxylase) that is approved in the US for the treatment of Cushing disease in patients where pituitary surgery is not an option or has not been curative, and in Europe and Japan for the treatment of endogenous Cushing syndrome.[26]Fleseriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing's disease: a guideline update. Lancet Diabetes Endocrinol. 2021 Dec;9(12):847-75.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8743006
http://www.ncbi.nlm.nih.gov/pubmed/34687601?tool=bestpractice.com
[102]Pivonello R, Fleseriu M, Newell-Price J, et al. Efficacy and safety of osilodrostat in patients with Cushing's disease (LINC 3): a multicentre phase III study with a double-blind, randomised withdrawal phase. Lancet Diabetes Endocrinol. 2020 Sep;8(9):748-61.
http://www.ncbi.nlm.nih.gov/pubmed/32730798?tool=bestpractice.com
Osilodrostat rapidly reduces urinary free cortisol with associated improvements in clinical signs of hypercortisolism, and is generally well tolerated.[102]Pivonello R, Fleseriu M, Newell-Price J, et al. Efficacy and safety of osilodrostat in patients with Cushing's disease (LINC 3): a multicentre phase III study with a double-blind, randomised withdrawal phase. Lancet Diabetes Endocrinol. 2020 Sep;8(9):748-61.
http://www.ncbi.nlm.nih.gov/pubmed/32730798?tool=bestpractice.com
There is a risk of hypocortisolism, hypokalemia, and QTc prolongation; careful monitoring for hyperandrogenism is needed in women.[26]Fleseriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing's disease: a guideline update. Lancet Diabetes Endocrinol. 2021 Dec;9(12):847-75.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8743006
http://www.ncbi.nlm.nih.gov/pubmed/34687601?tool=bestpractice.com
Ketoconazole has a relatively rapid onset of steroidogenesis inhibition. Ketoconazole may cause idiopathic severe liver injury and adrenal insufficiency.[93]Hinojosa-Amaya JM, Cuevas-Ramos D, Fleseriu M. Medical management of Cushing's syndrome: current and emerging treatments. Drugs. 2019 Jun;79(9):935-56.
http://www.ncbi.nlm.nih.gov/pubmed/31098899?tool=bestpractice.com
[103]Castinetti F, Guignat L, Giraud P, et al. Ketoconazole in Cushing's disease: is it worth a try? J Clin Endocrinol Metab. 2014 May;99(5):1623-30.
https://academic.oup.com/jcem/article/99/5/1623/2537349
http://www.ncbi.nlm.nih.gov/pubmed/24471573?tool=bestpractice.com
[104]US Food and Drug Administration. FDA drug safety communication: FDA warns that prescribing of Nizoral (ketoconazole) oral tablets for unapproved uses including skin and nail infections continues; linked to patient death. May 2016 [internet publication].
https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-warns-prescribing-nizoral-ketoconazole-oral-tablets-unapproved
Its use requires expert guidance and is contraindicated in patients with liver disease. If used, liver and adrenal function should be monitored before and during treatment.[104]US Food and Drug Administration. FDA drug safety communication: FDA warns that prescribing of Nizoral (ketoconazole) oral tablets for unapproved uses including skin and nail infections continues; linked to patient death. May 2016 [internet publication].
https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-warns-prescribing-nizoral-ketoconazole-oral-tablets-unapproved
Levoketoconazole is an adrenal steroidogenesis inhibitor that is approved for treatment in the US.[105]Fleseriu M, Castinetti F. Updates on the role of adrenal steroidogenesis inhibitors in Cushing's syndrome: a focus on novel therapies. Pituitary. 2016 Dec;19(6):643-53.
https://link.springer.com/article/10.1007/s11102-016-0742-1
http://www.ncbi.nlm.nih.gov/pubmed/27600150?tool=bestpractice.com
[106]Fleseriu M, Pivonello R, Elenkova A, et al. Efficacy and safety of levoketoconazole in the treatment of endogenous Cushing's syndrome (SONICS): a phase 3, multicentre, open-label, single-arm trial. Lancet Diabetes Endocrinol. 2019 Nov;7(11):855-65.
http://www.ncbi.nlm.nih.gov/pubmed/31542384?tool=bestpractice.com
[107]Zacharieva SZ, Pivonello R, Elenkova A, et al. MON-332 safety and efficacy of levoketoconazole in the treatment of endogenous Cushing’s syndrome (LOGICS): a double-blind, placebo-controlled, withdrawal study. J Endocr Soc. 2020 May 8;4(suppl 1):MON-332.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7208150
Metyrapone provides rapid onset of inhibition and can be obtained for compassionate use in the US.
Mitotane has adrenostatic and adrenolytic properties, but has delayed efficacy due to slow onset of action and a narrow therapeutic window.[93]Hinojosa-Amaya JM, Cuevas-Ramos D, Fleseriu M. Medical management of Cushing's syndrome: current and emerging treatments. Drugs. 2019 Jun;79(9):935-56.
http://www.ncbi.nlm.nih.gov/pubmed/31098899?tool=bestpractice.com
It is rarely used for Cushing syndrome due to causes other than adrenal carcinoma.
Etomidate is a potent adrenostatic agent with a rapid onset of action. It is used only in emergencies (e.g., hypercortisol-induced psychosis), and must be given intravenously.[93]Hinojosa-Amaya JM, Cuevas-Ramos D, Fleseriu M. Medical management of Cushing's syndrome: current and emerging treatments. Drugs. 2019 Jun;79(9):935-56.
http://www.ncbi.nlm.nih.gov/pubmed/31098899?tool=bestpractice.com
Mifepristone, a glucocorticoid receptor antagonist, blocks the effect of cortisol at the receptor level and should be considered in patients who have clinical and metabolic derangements of continued hypercortisolism with hyperglycemia and/or diabetes. The US Food and Drug Administration has approved mifepristone for the treatment of hyperglycemia associated with Cushing syndrome in patients with type 2 diabetes mellitus. Cortisol and ACTH levels may increase with the use of mifepristone due to feedback inhibition.[108]Fleseriu M, Findling JW, Koch CA, et al. Changes in plasma ACTH levels and corticotroph tumor size in patients with Cushing's disease during long-term treatment with the glucocorticoid receptor antagonist mifepristone. J Clin Endocrinol Metab. 2014 Oct;99(10):3718-27.
https://academic.oup.com/jcem/article/99/10/3718/2836437
http://www.ncbi.nlm.nih.gov/pubmed/25013998?tool=bestpractice.com
As such, cortisol levels should not be used to guide therapy in patients treated with mifepristone.[109]Fleseriu M, Biller BM, Findling JW, et al; SEISMIC Study Investigators. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing's syndrome. J Clin Endocrinol Metab. 2012 Jun;97(6):2039-49.
https://academic.oup.com/jcem/article/97/6/2039/2536684
http://www.ncbi.nlm.nih.gov/pubmed/22466348?tool=bestpractice.com
Bilateral adrenalectomy provides an immediate cure to any cause of endogenous hypercortisolism, at the expense of causing permanent adrenal insufficiency (requiring cortisol and mineralocorticoid replacement) and creating a risk of Nelson syndrome (corticotroph tumor growth after adrenalectomy). This progression can cause hyperpigmentation from excessive ACTH and intracranial compressive symptoms from growth of the tumor outside the sella. Newer laparoscopic methods of adrenalectomy allow for more rapid recovery and tolerability.[110]Neychev V, Steinberg SM, Yang L, et al. Long-term outcome of bilateral laparoscopic adrenalectomy measured by disease-specific questionnaire in a unique group of patients with Cushing's syndrome. Ann Surg Oncol. 2015 Dec;22 Suppl 3:S699-706.
http://www.ncbi.nlm.nih.gov/pubmed/25968622?tool=bestpractice.com
One meta-analysis of 37 studies (1320 patients, 82% with Cushing disease, 13% with ectopic Cushing syndrome, and 5% with primary adrenal hyperplasia) showed that bilateral adrenalectomy is relatively safe and provides adequate success.[111]Ritzel K, Beuschlein F, Mickisch A, et al. Clinical review: outcome of bilateral adrenalectomy in Cushing's syndrome: a systematic review. J Clin Endocrinol Metab. 2013 Oct;98(10):3939-48.
https://academic.oup.com/jcem/article/98/10/3939/2833849
http://www.ncbi.nlm.nih.gov/pubmed/23956347?tool=bestpractice.com
Although residual cortisol secretion due to accessory adrenal tissue or adrenal remnants was found in up to 34% of patients, less than 2% had a relapse of Cushing syndrome. Symptoms of hypercortisolism (e.g., hypertension, obesity, or depression) improved in the majority of the patients after bilateral adrenalectomy. The number of adrenal crises per 100 patient years was 9.3, and Nelson syndrome occurred in 21% of the patients. Excess mortality within the first year after surgery suggests that intensive clinical care for patients after bilateral adrenalectomy is warranted.
For replacement of non-glucocorticoid pituitary hormones following pituitary surgery (any combination of deficiencies may occur):[112]Fleseriu M, Hashim IA, Karavitaki N, et al. Hormonal replacement in hypopituitarism in adults: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016 Nov;101(11):3888-921.
https://academic.oup.com/jcem/article/101/11/3888/2764912
http://www.ncbi.nlm.nih.gov/pubmed/27736313?tool=bestpractice.com
Levothyroxine is used to achieve a free T4 in the upper half of the normal range. A thyroid-stimulating hormone should not be used to guide therapy.
Testosterone therapy is used to achieve a testosterone level in the normal range.
Women with an intact uterus taking estrogen replacement also need 10 days of progestin each month in addition to estrogen replacement therapy.
Decision to treat with growth hormone should be individualized for each patient based on symptoms, benefits, and risk of therapy. Dose titration should occur every month to achieve clinical response (i.e., energy level, sense of well-being, and lean body mass) and an insulin-like growth factor 1 (IGF-1) level in the age-adjusted mid- to upper-normal range.
Desmopressin is titrated to control symptomatic excessive urine output. This is based on patient preference. Serum sodium and symptoms are monitored periodically to evaluate adequacy of treatment.
ACTH-independent Cushing syndrome
The most common cause of adrenal cortisol overproduction is a unilateral autonomous adrenal adenoma. First-line therapy is almost always unilateral adrenalectomy of the affected adrenal gland. Laparoscopic adrenalectomy is the preferred method in most cases. Removal of the affected adrenal gland is curative in all patients with unilateral adrenal disease. Adrenalectomy has a beneficial effect on cardiovascular risk factors in patients with subclinical Cushing syndrome overall and compared with conservative management.[113]Bancos I, Alahdab F, Crowley RK, et al. Therapy of endocrine disease: improvement of cardiovascular risk factors after adrenalectomy in patients with adrenal tumors and subclinical Cushing's syndrome: a systematic review and meta-analysis. Eur J Endocrinol. 2016 Dec;175(6):R283-95.
https://eje.bioscientifica.com/view/journals/eje/175/6/R283.xml
http://www.ncbi.nlm.nih.gov/pubmed/27450696?tool=bestpractice.com
Rare causes of ACTH-independent disease generally cause bilateral adrenal disease from autonomous nodule formation or bilateral hyperplasia.[19]Duan K, Gomez Hernandez K, Mete O. Clinicopathological correlates of adrenal Cushing's syndrome. J Clin Pathol. 2015 Mar;68(3):175-86.
https://jcp.bmj.com/content/68/3/175.long
http://www.ncbi.nlm.nih.gov/pubmed/25425660?tool=bestpractice.com
[92]Feelders RA, Newell-Price J, Pivonello R, et al. Advances in the medical treatment of Cushing's syndrome. Lancet Diabetes Endocrinol. 2019 Apr;7(4):300-12.
http://www.ncbi.nlm.nih.gov/pubmed/30033041?tool=bestpractice.com
In these cases first-line therapy generally requires bilateral adrenalectomy. Steroidogenesis inhibitor therapy (osilodrostat, ketoconazole, levoketoconazole, metyrapone, mitotane, and etomidate), or a glucocorticoid receptor antagonist (mifepristone) can be used for patients who wish to avoid bilateral adrenalectomy.[19]Duan K, Gomez Hernandez K, Mete O. Clinicopathological correlates of adrenal Cushing's syndrome. J Clin Pathol. 2015 Mar;68(3):175-86.
https://jcp.bmj.com/content/68/3/175.long
http://www.ncbi.nlm.nih.gov/pubmed/25425660?tool=bestpractice.com
[93]Hinojosa-Amaya JM, Cuevas-Ramos D, Fleseriu M. Medical management of Cushing's syndrome: current and emerging treatments. Drugs. 2019 Jun;79(9):935-56.
http://www.ncbi.nlm.nih.gov/pubmed/31098899?tool=bestpractice.com
[114]Cohan P, East HE, Galati SJ, et al. Mifepristone treatment in four cases of primary bilateral macronodular adrenal hyperplasia (BMAH). J Clin Endocrinol Metab. 2019 Dec 1;104(12):6279-90.
https://academic.oup.com/jcem/article/104/12/6279/5491614
http://www.ncbi.nlm.nih.gov/pubmed/31112270?tool=bestpractice.com
Osilodrostat rapidly reduces urinary free cortisol with associated improvements in clinical signs of hypercortisolism, and is generally well tolerated.[26]Fleseriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing's disease: a guideline update. Lancet Diabetes Endocrinol. 2021 Dec;9(12):847-75.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8743006
http://www.ncbi.nlm.nih.gov/pubmed/34687601?tool=bestpractice.com
[102]Pivonello R, Fleseriu M, Newell-Price J, et al. Efficacy and safety of osilodrostat in patients with Cushing's disease (LINC 3): a multicentre phase III study with a double-blind, randomised withdrawal phase. Lancet Diabetes Endocrinol. 2020 Sep;8(9):748-61.
http://www.ncbi.nlm.nih.gov/pubmed/32730798?tool=bestpractice.com
Ketoconazole has a relatively rapid onset of steroidogenesis inhibition. Ketoconazole may cause severe liver injury and adrenal insufficiency. Its use requires expert guidance and is contraindicated in patients with liver disease. If used, liver and adrenal function should be monitored before and during treatment.[104]US Food and Drug Administration. FDA drug safety communication: FDA warns that prescribing of Nizoral (ketoconazole) oral tablets for unapproved uses including skin and nail infections continues; linked to patient death. May 2016 [internet publication].
https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-warns-prescribing-nizoral-ketoconazole-oral-tablets-unapproved
Levoketoconazole is an adrenal steroidogenesis inhibitor that is approved for treatment in the US.[105]Fleseriu M, Castinetti F. Updates on the role of adrenal steroidogenesis inhibitors in Cushing's syndrome: a focus on novel therapies. Pituitary. 2016 Dec;19(6):643-53.
https://link.springer.com/article/10.1007/s11102-016-0742-1
http://www.ncbi.nlm.nih.gov/pubmed/27600150?tool=bestpractice.com
[106]Fleseriu M, Pivonello R, Elenkova A, et al. Efficacy and safety of levoketoconazole in the treatment of endogenous Cushing's syndrome (SONICS): a phase 3, multicentre, open-label, single-arm trial. Lancet Diabetes Endocrinol. 2019 Nov;7(11):855-65.
http://www.ncbi.nlm.nih.gov/pubmed/31542384?tool=bestpractice.com
[107]Zacharieva SZ, Pivonello R, Elenkova A, et al. MON-332 safety and efficacy of levoketoconazole in the treatment of endogenous Cushing’s syndrome (LOGICS): a double-blind, placebo-controlled, withdrawal study. J Endocr Soc. 2020 May 8;4(suppl 1):MON-332.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7208150
Metyrapone provides rapid onset of inhibition and can be obtained for compassionate use in the US. Mitotane has slow onset of action, has a narrow therapeutic window, and is generally only used in adrenal carcinoma.[72]Pivonello R, De Leo M, Cozzolino A, et al. The treatment of Cushing's disease. Endocr Rev. 2015 Aug;36(4):385-486.
https://academic.oup.com/edrv/article/36/4/385/2354703
http://www.ncbi.nlm.nih.gov/pubmed/26067718?tool=bestpractice.com
Etomidate, used only in emergencies, has rapid onset but must be given intravenously.[72]Pivonello R, De Leo M, Cozzolino A, et al. The treatment of Cushing's disease. Endocr Rev. 2015 Aug;36(4):385-486.
https://academic.oup.com/edrv/article/36/4/385/2354703
http://www.ncbi.nlm.nih.gov/pubmed/26067718?tool=bestpractice.com
Mifepristone blocks cortisol action, resulting in attenuation of cortisol effects.
Adrenal carcinoma is an exceedingly rare cause of ACTH-independent Cushing syndrome. First-line therapy in many patients is surgical resection; however, at the time of diagnosis the disease has often progressed beyond the point where surgical therapy is effective. The effectiveness of chemotherapy and adjunctive therapies in both early- and late-stage disease has shown mixed results in clinical trials; however, patients should be considered for treatment with mitotane and enrollment in clinical trials (if available).[10]Fassnacht M, Dekkers OM, Else T, et al. European Society of Endocrinology clinical practice guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol. 2018 Oct 1;179(4):G1-46.
https://eje.bioscientifica.com/view/journals/eje/179/4/EJE-18-0608.xml
http://www.ncbi.nlm.nih.gov/pubmed/30299884?tool=bestpractice.com
Ectopic ACTH syndrome
The optimal first-line therapy involves locating and surgically resecting the ACTH-producing tumor. Not infrequently, complete resection of these tumors is not possible. Where surgical resection is not possible, second-line therapies include a glucocorticoid receptor antagonist (mifepristone), steroidogenesis inhibitor therapy (osilodrostat, ketoconazole, levoketoconazole, metyrapone, mitotane, and etomidate), or bilateral adrenalectomy.[92]Feelders RA, Newell-Price J, Pivonello R, et al. Advances in the medical treatment of Cushing's syndrome. Lancet Diabetes Endocrinol. 2019 Apr;7(4):300-12.
http://www.ncbi.nlm.nih.gov/pubmed/30033041?tool=bestpractice.com
Bilateral adrenalectomy is definitive therapy, but patients with ectopic ACTH-producing tumors may have extremely severe hypercortisolism, and require reduction in cortisol before proceeding to surgery. In these cases, mifepristone or steroidogenesis inhibitor therapy can be used to block cortisol action or lower cortisol levels in preparation for bilateral adrenalectomy. Osilodrostat rapidly reduces urinary free cortisol with associated improvements in clinical signs of hypercortisolism, and is generally well tolerated.[26]Fleseriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing's disease: a guideline update. Lancet Diabetes Endocrinol. 2021 Dec;9(12):847-75.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8743006
http://www.ncbi.nlm.nih.gov/pubmed/34687601?tool=bestpractice.com
[102]Pivonello R, Fleseriu M, Newell-Price J, et al. Efficacy and safety of osilodrostat in patients with Cushing's disease (LINC 3): a multicentre phase III study with a double-blind, randomised withdrawal phase. Lancet Diabetes Endocrinol. 2020 Sep;8(9):748-61.
http://www.ncbi.nlm.nih.gov/pubmed/32730798?tool=bestpractice.com
Ketoconazole has a relatively rapid onset of steroidogenesis inhibition. Ketoconazole may cause severe liver injury and adrenal insufficiency. Its use requires expert guidance and is contraindicated in patients with liver disease. If used, liver and adrenal function should be monitored before and during treatment.[104]US Food and Drug Administration. FDA drug safety communication: FDA warns that prescribing of Nizoral (ketoconazole) oral tablets for unapproved uses including skin and nail infections continues; linked to patient death. May 2016 [internet publication].
https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-warns-prescribing-nizoral-ketoconazole-oral-tablets-unapproved
Levoketoconazole is an adrenal steroidogenesis inhibitor that is approved for treatment in the US.[105]Fleseriu M, Castinetti F. Updates on the role of adrenal steroidogenesis inhibitors in Cushing's syndrome: a focus on novel therapies. Pituitary. 2016 Dec;19(6):643-53.
https://link.springer.com/article/10.1007/s11102-016-0742-1
http://www.ncbi.nlm.nih.gov/pubmed/27600150?tool=bestpractice.com
[106]Fleseriu M, Pivonello R, Elenkova A, et al. Efficacy and safety of levoketoconazole in the treatment of endogenous Cushing's syndrome (SONICS): a phase 3, multicentre, open-label, single-arm trial. Lancet Diabetes Endocrinol. 2019 Nov;7(11):855-65.
http://www.ncbi.nlm.nih.gov/pubmed/31542384?tool=bestpractice.com
[107]Zacharieva SZ, Pivonello R, Elenkova A, et al. MON-332 safety and efficacy of levoketoconazole in the treatment of endogenous Cushing’s syndrome (LOGICS): a double-blind, placebo-controlled, withdrawal study. J Endocr Soc. 2020 May 8;4(suppl 1):MON-332.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7208150
Metyrapone provides rapid onset of inhibition and can be obtained for compassionate use in the US. Mitotane has slow onset of action, has a narrow therapeutic window, and is generally only used in adrenal carcinoma.[72]Pivonello R, De Leo M, Cozzolino A, et al. The treatment of Cushing's disease. Endocr Rev. 2015 Aug;36(4):385-486.
https://academic.oup.com/edrv/article/36/4/385/2354703
http://www.ncbi.nlm.nih.gov/pubmed/26067718?tool=bestpractice.com
Etomidate, used only in emergencies, has rapid onset but must be given intravenously.[72]Pivonello R, De Leo M, Cozzolino A, et al. The treatment of Cushing's disease. Endocr Rev. 2015 Aug;36(4):385-486.
https://academic.oup.com/edrv/article/36/4/385/2354703
http://www.ncbi.nlm.nih.gov/pubmed/26067718?tool=bestpractice.com
Mifepristone blocks cortisol action, resulting in attenuation of cortisol effects.