Etiology
Exogenous corticosteroid exposure is the most common cause of Cushing syndrome.
The majority (70% to 80%) of patients with endogenous Cushing syndrome have adrenocorticotropic hormone (ACTH)-secreting pituitary adenomas; however, only 10% of all pituitary adenomas secrete excessive ACTH.[8][16] Silent corticotroph adenomas are immunopositive for ACTH, but clinically present as nonfunctioning adenomas. However, large adenomas can rarely become ACTH secreting and cause Cushing disease.[17] A new gene, ubiquitin-specific protease 8, has been found to be frequently mutated in Cushing disease.[18]
About 10% of patients with endogenous Cushing syndrome have adrenal adenomas with unregulated secretion of cortisol, but only 5% of all adrenal adenomas develop autonomous cortisol secretion.[8][14] Mutations in the cAMP/PKA pathway are the cause of most cases of primary adrenal hypercortisolism.[19] No specific exposures or modifiable factors have been identified that cause endogenous hypercortisolism. The etiology of pituitary overproduction of ACTH and adrenal adenoma overproduction of cortisol is poorly understood.
Only about 1% of patients with Cushing syndrome have adrenal carcinoma. On the other hand, adrenal overproduction of cortisol is seen in 50% to 60% of adrenal carcinomas, resulting in ACTH-independent Cushing syndrome.[10]
Pathophysiology
The clinical manifestations result from excess tissue exposure to cortisol. The degree to which symptoms manifest is largely, if not entirely, based on the degree of cortisol excess. Patients with mild to moderate hypercortisolism generally have a less prominent phenotype with glucose intolerance, dyslipidemia, metabolic bone disease, and abnormal weight gain, but are difficult to differentiate from other patients with the metabolic syndrome. As the hypercortisolism increases, physical features worsen with striae, supraclavicular fat pads, and proximal muscle weakness developing. Ectopic adrenocorticotropic hormone (ACTH) secretion from neuroendocrine tumors may manifest as more-severe cases with the abrupt presentation of symptoms and greatly elevated cortisol and ACTH. These patients may also have severe muscle weakness and weight loss.
Classification
Etiologies of hypercortisolism
Adrenocorticotropic hormone (ACTH)-dependent
Caused by conditions that have high or inappropriately normal ACTH levels stimulating adrenal cortisol overproduction.
ACTH-secreting pituitary adenomas (Cushing disease) and ectopic ACTH-secreting tumors are two forms of ACTH-dependent disease. Ectopic ACTH-secreting tumors are typically of bronchogenic or neuroendocrine origin.
Ectopic corticotropin-releasing hormone can be considered in this category, but is extremely rare and is not specifically discussed in detail here.
ACTH-independent
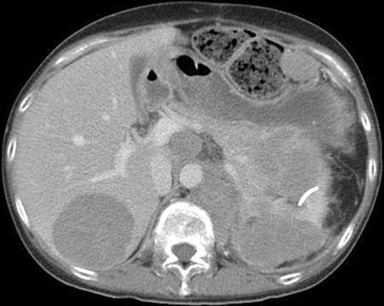
ACTH-independent Cushing syndrome is caused by excessive cortisol secretion by the adrenal glands despite a suppressed ACTH level. Included in this category are adrenal adenomas, bilateral adrenal hyperplasia, and, rarely, adrenal carcinoma. Adrenal carcinoma is extremely rare and typically presents as a large (>5 cm) and rapidly growing adrenal mass.[Figure caption and citation for the preceding image starts]: Abdominal computed scan showing adrenocortical tumor infiltrating the pancreas and left kidney, and metastasized to the liver, spleen, and central nodesFrom BMJ Case Reports 2010; doi:10.1136/bcr.07.2009.2100 [Citation ends].
ACTH-independent bilateral macronodular adrenal hyperplasia (AIMAH) is a rare form of bilateral nodular adrenal disease. The pathophysiology of AIMAH is aberrant receptor stimulation. At least 10 different aberrant receptors causing AIMAH have been identified. They include gastric inhibitory polypeptide receptor, beta-adrenergic receptors, vasopressin receptor, serotonin receptor, angiotensin II receptor, luteinizing hormone/human chorionic gonadotropin receptor, and leptin receptor.[2] Stimulation of these receptors leads to inappropriate growth of bilateral, large, monoclonal and polyclonal nodules.[3][4] Clinically, AIMAH presents most commonly in the fifth and sixth decades of life with excess glucocorticoid and mineralocorticoid production.[5] On imaging, bilaterally massively enlarged adrenal glands are seen.
Primary pigmented nodular adrenocortical disease (PPNAD), often part of Carney complex (a multiple endocrine neoplasia syndrome characterized by lentigines, cardiac and cutaneous myxomas, and endocrine tumors; may be inherited in an autosomal-dominant manner), is a rare condition caused by numerous small, autonomously functioning adrenal nodules ranging in size from microscopic to 1 cm. On pathologic exam the nodules are darkly pigmented, and the intervening adrenal cortex is atrophic. Mutations in the protein kinase A type 1 alpha regulatory subunit (PRKAR1A) are believed to be the causative mutations. PPNAD has a bimodal age of distribution, with most cases being diagnosed in the second and third decades of life.[6] Patients with PPNAD tend to be rather lean, and present without the typical central obesity seen in other causes of Cushing syndrome. Severe osteoporosis, short stature, and severe muscle wasting are common presenting features of patients with PPNAD.[7]
Exogenous
Patients taking exogenous corticosteroids for any reason may develop features of Cushing syndrome. When high-dose glucocorticosteroids are stopped, patients can develop adrenal insufficiency despite a clinical phenotype of Cushing.
Use of this content is subject to our disclaimer