History
A pheochromocytoma should be suspected in any patient who presents with the classic triad of symptoms:[1]Neumann HPH, Young WF Jr, Eng C. Pheochromocytoma and paraganglioma. N Engl J Med. 2019 Aug 8;381(6):552-65.
http://www.ncbi.nlm.nih.gov/pubmed/31390501?tool=bestpractice.com
[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
[3]Martucci VL, Pacak K. Pheochromocytoma and paraganglioma: diagnosis, genetics, management, and treatment. Curr Probl Cancer. 2014 Jan-Feb;38(1):7-41.
https://www.cpcancer.com/article/S0147-0272(14)00002-6/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/24636754?tool=bestpractice.com
[43]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: neuroendocrine and adrenal tumors [internet publication].
https://www.nccn.org/guidelines/category_1
Palpitations
Headaches
Diaphoresis.
Episodic spells of the symptoms are characteristic; they can vary in duration from seconds to hours and typically get worse with time as the tumor enlarges.
Other suggestive features:
Inquiring about the family history is vital. Up to 40% of pheochromocytoma and paraganglioma (PPGL) cases are a manifestation of a hereditary syndrome (e.g., multiple endocrine neoplasia [MEN] syndrome type 2A and B; Von Hippel-Lindau [VHL] disease; neurofibromatosis type 1 [NF1]).[1]Neumann HPH, Young WF Jr, Eng C. Pheochromocytoma and paraganglioma. N Engl J Med. 2019 Aug 8;381(6):552-65.
http://www.ncbi.nlm.nih.gov/pubmed/31390501?tool=bestpractice.com
[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
[3]Martucci VL, Pacak K. Pheochromocytoma and paraganglioma: diagnosis, genetics, management, and treatment. Curr Probl Cancer. 2014 Jan-Feb;38(1):7-41.
https://www.cpcancer.com/article/S0147-0272(14)00002-6/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/24636754?tool=bestpractice.com
Germline mutations in the succinate dehydrogenase [SDH] subunit B, C, and D genes, or a personal history of a pheochromocytoma, increase risk.[35]Amar L, Pacak K, Steichen O, et al. International consensus on initial screening and follow-up of asymptomatic SDHx mutation carriers. Nat Rev Endocrinol. 2021 Jul;17(7):435-44.
https://www.nature.com/articles/s41574-021-00492-3
http://www.ncbi.nlm.nih.gov/pubmed/34021277?tool=bestpractice.com
[36]Andrews KA, Ascher DB, Pires DEV, et al. Tumour risks and genotype-phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD. J Med Genet. 2018 Jun;55(6):384-94.
https://jmg.bmj.com/content/55/6/384
http://www.ncbi.nlm.nih.gov/pubmed/29386252?tool=bestpractice.com
Clinical presentation of pheochromocytoma can vary widely and 10% to 15% of cases can be completely asymptomatic with the tumor discovered incidentally during abdominal investigation for other reasons.[44]Rogowski-Lehmann N, Geroula A, Prejbisz A, et al. Missed clinical clues in patients with pheochromocytoma/paraganglioma discovered by imaging. Endocr Connect. 2018 Sep 1 [Epub ahead of print].
https://www.doi.org/10.1530/EC-18-0318
http://www.ncbi.nlm.nih.gov/pubmed/30352425?tool=bestpractice.com
Approximately 3% to 7% of incidentally discovered adrenal masses are diagnosed as pheochromocytoma.[44]Rogowski-Lehmann N, Geroula A, Prejbisz A, et al. Missed clinical clues in patients with pheochromocytoma/paraganglioma discovered by imaging. Endocr Connect. 2018 Sep 1 [Epub ahead of print].
https://www.doi.org/10.1530/EC-18-0318
http://www.ncbi.nlm.nih.gov/pubmed/30352425?tool=bestpractice.com
It is recommended that all patients with such masses should undergo biochemical evaluation.[45]Fassnacht M, Arlt W, Bancos I, et al. Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol. 2016 Aug;175(2):G1-G34.
https://www.doi.org/10.1530/EJE-16-0467
http://www.ncbi.nlm.nih.gov/pubmed/27390021?tool=bestpractice.com
[46]Zeiger MA, Thompson GB, Duh QY, et al. American Association of Clinical Endocrinologists and American Association of Endocrine Surgeons medical guidelines for the management of adrenal incidentalomas: executive summary of recommendations. Endocr Pract. 2009 Jul-Aug;15(5):450-3.
http://www.ncbi.nlm.nih.gov/pubmed/19632968?tool=bestpractice.com
Physical exam findings
Hypertension is the principal sign on examination.[47]Zuber SM, Kantorovich V, Pacak K. Hypertension in pheochromocytoma: characteristics and treatment. Endocrinol Metab Clin North Am. 2011 Jun;40(2):295-311.
http://www.ncbi.nlm.nih.gov/pubmed/21565668?tool=bestpractice.com
Patients often present with accelerated hypertension or hypertension refractory to multiple drug regimens. In about 48% of cases the hypertension is paroxysmal or labile in nature.[47]Zuber SM, Kantorovich V, Pacak K. Hypertension in pheochromocytoma: characteristics and treatment. Endocrinol Metab Clin North Am. 2011 Jun;40(2):295-311.
http://www.ncbi.nlm.nih.gov/pubmed/21565668?tool=bestpractice.com
Pheochromocytomas may present with life-threatening acute hypertensive emergencies (e.g., encephalopathy), as well as clinical consequences of long-lasting hypertension (e.g., hypertensive retinopathy, proteinuria, cardiomyopathies, or arrhythmias).[47]Zuber SM, Kantorovich V, Pacak K. Hypertension in pheochromocytoma: characteristics and treatment. Endocrinol Metab Clin North Am. 2011 Jun;40(2):295-311.
http://www.ncbi.nlm.nih.gov/pubmed/21565668?tool=bestpractice.com
A hypertensive crisis can be triggered by medications, intravenous contrast, surgery, or even exercise. Postural hypotension may be a feature due to volume contraction. Other signs associated with pheochromocytomas include abdominal masses, tachycardia, pallor, or tremors.[1]Neumann HPH, Young WF Jr, Eng C. Pheochromocytoma and paraganglioma. N Engl J Med. 2019 Aug 8;381(6):552-65.
http://www.ncbi.nlm.nih.gov/pubmed/31390501?tool=bestpractice.com
[3]Martucci VL, Pacak K. Pheochromocytoma and paraganglioma: diagnosis, genetics, management, and treatment. Curr Probl Cancer. 2014 Jan-Feb;38(1):7-41.
https://www.cpcancer.com/article/S0147-0272(14)00002-6/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/24636754?tool=bestpractice.com
Laboratory evaluation
All patients with palpitations, headaches, and diaphoresis should be investigated, whether they have hypertension or not.[27]Fishbein L, Del Rivero J, Else T, et al. The North American Neuroendocrine Tumor Society consensus guidelines for surveillance and management of metastatic and/or unresectable pheochromocytoma and paraganglioma. Pancreas. 2021 Apr 1;50(4):469-93.
https://nanets.net/images/2021/2021_NANETS_Consensus_Guidelines_for_Surveillance_and_Management_of_Metastatic_and_or_Unresectable_Pheochromocytoma_and_Paraganglioma.pdf
http://www.ncbi.nlm.nih.gov/pubmed/33939658?tool=bestpractice.com
Investigations should be carried out in any patient with hereditary risk that predisposes to pheochromocytoma development, such as MEN2.[27]Fishbein L, Del Rivero J, Else T, et al. The North American Neuroendocrine Tumor Society consensus guidelines for surveillance and management of metastatic and/or unresectable pheochromocytoma and paraganglioma. Pancreas. 2021 Apr 1;50(4):469-93.
https://nanets.net/images/2021/2021_NANETS_Consensus_Guidelines_for_Surveillance_and_Management_of_Metastatic_and_or_Unresectable_Pheochromocytoma_and_Paraganglioma.pdf
http://www.ncbi.nlm.nih.gov/pubmed/33939658?tool=bestpractice.com
Biochemical tests
Measurement of plasma free metanephrines, or 24-hour urine fractionated metanephrines and normetanephrines, is recommended in patients with suspected pheochromocytoma.[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
[27]Fishbein L, Del Rivero J, Else T, et al. The North American Neuroendocrine Tumor Society consensus guidelines for surveillance and management of metastatic and/or unresectable pheochromocytoma and paraganglioma. Pancreas. 2021 Apr 1;50(4):469-93.
https://nanets.net/images/2021/2021_NANETS_Consensus_Guidelines_for_Surveillance_and_Management_of_Metastatic_and_or_Unresectable_Pheochromocytoma_and_Paraganglioma.pdf
http://www.ncbi.nlm.nih.gov/pubmed/33939658?tool=bestpractice.com
[43]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: neuroendocrine and adrenal tumors [internet publication].
https://www.nccn.org/guidelines/category_1
Elevations 3 times above the upper limit of normal are diagnostic.[43]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: neuroendocrine and adrenal tumors [internet publication].
https://www.nccn.org/guidelines/category_1
Blood sampling should be performed in the supine position.[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
[26]Granberg D, Juhlin CC, Falhammar H. Metastatic pheochromocytomas and abdominal paragangliomas. J Clin Endocrinol Metab. 2021 Apr 23;106(5):e1937-52.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8063253
http://www.ncbi.nlm.nih.gov/pubmed/33462603?tool=bestpractice.com
Some drugs may interfere with testing results (e.g., acetaminophen, buspirone, cocaine, labetalol, levodopa, methyldopa, monoamine oxidase inhibitors [MAOIs], phenoxybenzamine, sotalol, sulfasalazine, sympathomimetics, tricyclic antidepressants); review patient medications accordingly.[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
[43]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: neuroendocrine and adrenal tumors [internet publication].
https://www.nccn.org/guidelines/category_1
Note that urine or plasma catecholamines are no longer routinely recommended for the evaluation of pheochromocytoma.[27]Fishbein L, Del Rivero J, Else T, et al. The North American Neuroendocrine Tumor Society consensus guidelines for surveillance and management of metastatic and/or unresectable pheochromocytoma and paraganglioma. Pancreas. 2021 Apr 1;50(4):469-93.
https://nanets.net/images/2021/2021_NANETS_Consensus_Guidelines_for_Surveillance_and_Management_of_Metastatic_and_or_Unresectable_Pheochromocytoma_and_Paraganglioma.pdf
http://www.ncbi.nlm.nih.gov/pubmed/33939658?tool=bestpractice.com
[43]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: neuroendocrine and adrenal tumors [internet publication].
https://www.nccn.org/guidelines/category_1
[48]American Society for Clinical Pathology. Thirty five things physicians and patients should question. Choosing Wisely, an initiative of the ABIM Foundation. 2019 [internet publication].
https://web.archive.org/web/20221207180203/https://www.choosingwisely.org/societies/endocrine-society
[49]Nölting S, Bechmann N, Taieb D, et al. Personalized management of pheochromocytoma and paraganglioma. Endocr Rev. 2022 Mar 9;43(2):199-239.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8905338
http://www.ncbi.nlm.nih.gov/pubmed/34147030?tool=bestpractice.com
A clonidine suppression test can be used to discriminate patients with mildly elevated test results for plasma normetanephrine (attributable to increased sympathetic activity) from those with elevated test results due to a PPGL.[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
Chromogranin A may be elevated in patients with a neuroendocrine tumor; chromogranin A plus urinary fractionated metanephrines have been suggested as follow-up tests for elevations of plasma metanephrines.[50]Algeciras-Schimnich A, Preissner CM, Young WF Jr, et al. Plasma chromogranin A or urine fractionated metanephrines follow-up testing improves the diagnostic accuracy of plasma fractionated metanephrines for pheochromocytoma. J Clin Endocrinol Metab. 2008 Jan;93(1):91-5.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2729153
http://www.ncbi.nlm.nih.gov/pubmed/17940110?tool=bestpractice.com
Imaging studies
Localization studies should only be undertaken after a biochemical abnormality is demonstrated.
CT imaging is recommended given its excellent spatial resolution in the abdomen; MRI is an option for patients in whom CT imaging is contraindicated.[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
[16]Chen H, Sippel RS, O'Dorisio MS, et al. The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010 Aug;39(6):775-83.
https://www.doi.org/10.1097/MPA.0b013e3181ebb4f0
http://www.ncbi.nlm.nih.gov/pubmed/20664475?tool=bestpractice.com
Multiphasic abdomen/pelvis CT or MRI, 18F-fluoro-2 deoxy-D-glucose (18F-FDG) PET/CT, or I-123 metaiodobenzylguanidine (MIBG) scintigraphy are performed, as appropriate, in the presence of metastatic or multifocal disease.[43]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: neuroendocrine and adrenal tumors [internet publication].
https://www.nccn.org/guidelines/category_1
18F-FDG PET/CT is preferred to MIBG scintigraphy in this scenario.[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
[43]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: neuroendocrine and adrenal tumors [internet publication].
https://www.nccn.org/guidelines/category_1
[Figure caption and citation for the preceding image starts]: Abdominal CT scan with mass in the left adrenal gland, compatible with a pheochromocytomaAlface MM et al. BMJ Case Rep. 2015 Aug 4;2015:bcr2015211184; used with permission [Citation ends].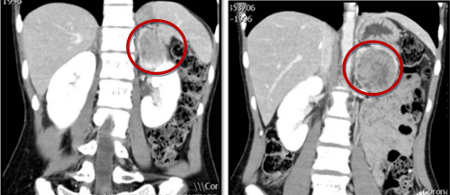 [Figure caption and citation for the preceding image starts]: Metaiodobenzylguanidine (MIBG) scintigraphy identified hyperfixation in the left adrenal gland compatible with pheochromocytomaAlface MM et al. BMJ Case Rep. 2015 Aug 4;2015:bcr2015211184; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Metaiodobenzylguanidine (MIBG) scintigraphy identified hyperfixation in the left adrenal gland compatible with pheochromocytomaAlface MM et al. BMJ Case Rep. 2015 Aug 4;2015:bcr2015211184; used with permission [Citation ends].
Genetic testing
All patients with pheochromocytomas should undergo genetic testing to identify potential hereditary tumor disorders that would necessitate more detailed evaluation and follow-up.[1]Neumann HPH, Young WF Jr, Eng C. Pheochromocytoma and paraganglioma. N Engl J Med. 2019 Aug 8;381(6):552-65.
http://www.ncbi.nlm.nih.gov/pubmed/31390501?tool=bestpractice.com
[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
[43]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: neuroendocrine and adrenal tumors [internet publication].
https://www.nccn.org/guidelines/category_1
Patient engagement in a shared decision-making process is essential.[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
[43]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: neuroendocrine and adrenal tumors [internet publication].
https://www.nccn.org/guidelines/category_1
Genetic testing may be considered in patients with pheochromocytoma with:[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
Positive family history (premised upon pedigree or identification of a PPGL-susceptibility gene mutation)
Syndromic features
Multifocal, bilateral, or metastatic disease
Targeted germline mutation testing (e.g, multiple endocrine neoplasia type 2 [MEN2], Von Hippel-Lindau syndrome [VHL], and neurofibromatosis type 1 [NF1]) is recommended in patients with positive family history or syndromic presentation.[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
Patients with metastatic disease should undergo testing for SDHB mutations.