Etiology
Lesch-Nyhan disease (LND) is caused by a mutation in the gene encoding hypoxanthine-guanine phosphoribosyltransferase (HPRT) on the long arm of the X-chromosome, at Xq26-q27.[10] The mutations are heterogeneous, including point mutations and other substitutions, deletions, and insertions.[11][12] It is generally believed that the majority of mutations occur de novo, because classic LND males do not reproduce. Mutations causing disease appear throughout the HPRT gene, with some minor mutational hot spots. Genotype-phenotype correlations do not indicate that specific disease features are associated with specific mutation sites. In general, however, less severe clinical manifestations (Lesch-Nyhan variants [LNV]) result from mutations predicted to allow some degree of residual enzyme function.[11][12] GeneReviews: Lesch-Nyhan syndrome Opens in new window
Pathophysiology
Hypoxanthine-guanine phosphoribosyltransferase (HPRT) mediates the recycling of hypoxanthine and guanine into their respective nucleotide pools. In the absence of HPRT, hypoxanthine and guanine are not recycled, but degraded to uric acid. The reduced purine salvage, together with an accompanying activation of de novo purine synthesis, causes marked overproduction of uric acid.[3]Despite this overproduction, efficient renal clearance limits average serum uric acid levels, which are typically increased less than two fold. The total renal uric acid excretion in classic Lesch-Nyhan disease (LND) is typically about four times that of controls. Chronic serum urate values >7.0 mg/dL are associated with an increased risk of deposition of urate crystals in joints, kidneys, and subcutaneous tissues.[13] If untreated, this may cause gouty arthritis, nephrolithiasis, and subcutaneous tophi.
LND is accompanied by a severe decrease in brain dopamine content in the basal ganglia, which is thought to be an important determinant of the neurobehavioral features, including the hyperkinetic motor disorder, attentional deficits, and abnormal behavior.[4][14][15] In addition, LND and Lesch-Nyhan variants (LNV) are associated with extensive abnormalities of gray matter and white matter that may also provide important clues to neural substrates of the phenotype.[16][17] The exact relation between HPRT deficiency and dopamine dysfunction in LND remains unclear. However, evidence suggests an important relationship between purine recycling pathways and the neurochemical integrity of the dopaminergic phenotype, and a role for HPRT in neurodevelopmental processes has been implied.[18][19][20] Despite the dopamine deficiency, the effect of dopamine replacement therapy is inconsistent and generally unhelpful, presumably because of the development of dopamine receptor super-sensitivity and other neuroplastic changes.[3][21] Some patients also develop spasticity and hyperreflexia, indicative of dysfunction of the corticospinal motor system which may result from myelopathy caused by chronic forceful involuntary movements of the neck.[22]
GeneReviews: Lesch-Nyhan syndrome
Opens in new window[Figure caption and citation for the preceding image starts]: The role of hypoxanthine-guanine phosphoribosyltransferase (HPRT) in the grand scheme of purine metabolismCreated by J.E. Visser, MD, PhD and H.A. Jinnah, MD, PhD; used with permission [Citation ends].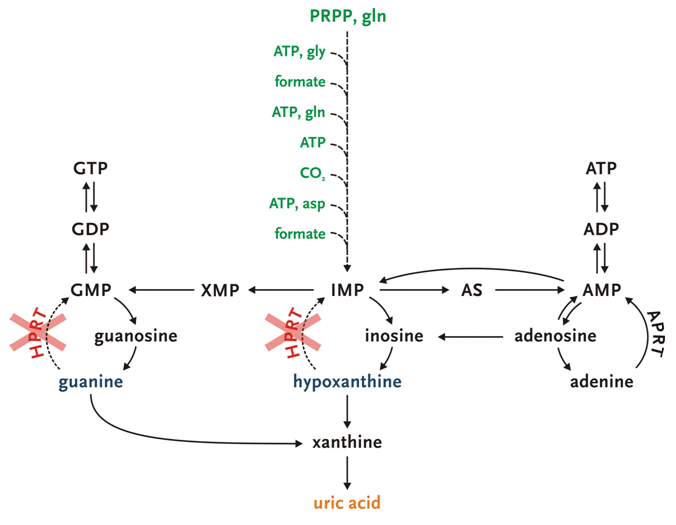 [Figure caption and citation for the preceding image starts]: Uric acid levels in patients with classic Lesch-Nyhan disease (LND), patients with Lesch-Nyhan variants (LNV), and healthy controls. SD, standard deviation; HRH: hypoxanthine-guanine phosphoribosyltransferase (HPRT)-related hyperuricemia; HRD: HPRT-related neurologic diseaseFrom the collection of J.E. Visser, MD, PhD and H.A. Jinnah, MD, PhD; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Uric acid levels in patients with classic Lesch-Nyhan disease (LND), patients with Lesch-Nyhan variants (LNV), and healthy controls. SD, standard deviation; HRH: hypoxanthine-guanine phosphoribosyltransferase (HPRT)-related hyperuricemia; HRD: HPRT-related neurologic diseaseFrom the collection of J.E. Visser, MD, PhD and H.A. Jinnah, MD, PhD; used with permission [Citation ends].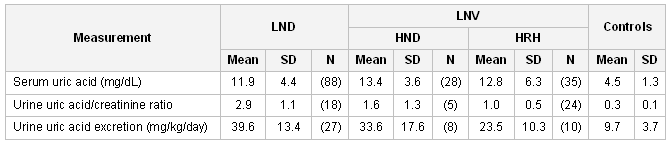
Classification
Lesch-Nyhan disease (LND), Lesch-Nyhan variants (LNV): HPRT-related neurologic disease and HPRT-related hyperuricemia[3][5][6]
HPRT deficiency is associated with a phenotypic continuum, where the occurrence and severity of clinical features is dependent on the residual enzyme activity.
Classic LND patients, with virtually no enzyme activity, have the full phenotype: hyperuricemia, neurologic dysfunction, cognitive deficits, and abnormal behavior including self-injury.
LNV, with some residual HPRT activity, have a partial phenotype: they do not perform self-injurious behavior, they may present with hyperuricemia with varying degrees of motor or cognitive dysfunction, or they may have hyperuricemia alone.[5] LNV are typically subdivided into:
HPRT-related neurologic disease (HND): hyperuricemia and some degree of neurologic dysfunction and/or cognitive deficits.
HPRT-related hyperuricemia (HRH): hyperuricemia alone, no neurologic dysfunction.
The eponym Kelley-Seegmiller syndrome has also been applied to LNV.[7] However, this term is best avoided; its definition is unclear because it has variously been used to refer to all variants or only to those with hyperuricemia alone. Moreover, it suggests another disease entity, but it is merely a variant of LND.[Figure caption and citation for the preceding image starts]: Phenotypic spectrum associated with hypoxanthine-guanine phosphoribosyltransferase (HPRT) deficiency. HND: HPRT-related neurologic disease; LND: Lesch-Nyhan diseaseFrom the collection of J.E. Visser, MD, PhD and H.A. Jinnah, MD, PhD; used with permission [Citation ends].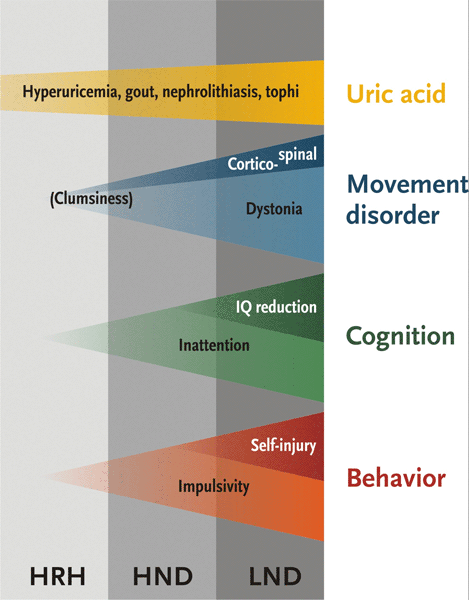
Use of this content is subject to our disclaimer