Approach
Girls who present before 8 years and boys who present before 9 years with early signs of puberty must be evaluated. The examining clinician should be well versed with the stages of normal puberty in order to know who to test and treat. A careful history and clinical examination should be performed. An accurate height, pubertal staging, and bone age should be recorded. Follow-up of height velocity and pubertal progress is most important for differentiating various etiologies and deciding the need for therapeutic intervention. Many patients do not require extensive investigations, although a sinister underlying cause such as a tumor should always be considered and excluded.
History
Assessment should initially include a detailed history for the onset of secondary sexual characteristics, the rate of pubertal progression, and, in girls, onset of menarche. An accurate history should be obtained about recent growth and if the child is outgrowing clothes very rapidly. Parental heights and age at onset of puberty, and menarche in the mother, should also be obtained. Social history should include any history of adoption.[35][36] Child abuse, particularly sexual abuse, is a risk factor for early pubertal development.[37] A full medication history should be taken to rule out exogenous drug ingestion.[22] History should be directed depending on whether puberty is consonant or disconsonant (i.e., if the pattern of endocrine change is the same as in normal puberty or not).
Normal pubertal development
Pubertal development and progress are best ascertained by Tanner staging.[1][2]
The first sign of puberty in boys is an increase in the size of the testes. This is followed by penile and scrotal changes. Testicular size is documented as a measurement of the longest axis or by the testicular volume using the Prader orchidometer. Volume of 4 mL or length of 2.5 cm (1 inch) defines the onset of puberty. Presence of axillary hair and other changes such as that of the voice and an increase in growth velocity only occur in mid to late puberty. Facial hair does not appear until late puberty.
[Figure caption and citation for the preceding image starts]: Prader orchidometerCreated by BMJ Knowledge Centre [Citation ends].
 [Figure caption and citation for the preceding image starts]: Method of comparing testicular size using the Prader orchidometerFrom the collection of Dr A. Mehta; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Method of comparing testicular size using the Prader orchidometerFrom the collection of Dr A. Mehta; used with permission [Citation ends].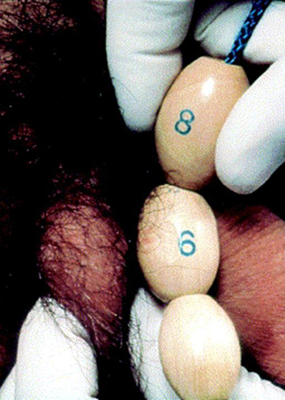
The first demonstrable sign of puberty in females is breast development. Caution must be exercised in examination of breast tissue in obese girls as simple fat may be mistaken for breast tissue. With true breast development, a small breast bud should be palpable behind the nipple. Pubic and axillary hair, acne, and body odor develop as a result of androgens secreted from the adrenal gland. Peak growth velocity occurs in Tanner stage 3 and menarche typically occurs at Tanner stage 4 breast development.
[Figure caption and citation for the preceding image starts]: Tanner staging: A, genital rating standards in boys; B, pubic hair rating standards in boys; C, breast rating standards in girls; D, pubic hair rating standards in girlsAdapted from Marshall WA, Tanner JM. Arch Dis Child. 1970;45:13-23; Marshall WA, Tanner JM. Arch Dis Child. 1969;44:291-303 [Citation ends].

Consonant or central precocious puberty (CPP)
Consonant pubertal development suggests a central cause. The hypothalamo-pituitary-gonadal axis is prematurely activated. In females, CPP is typically idiopathic (97% of cases in girls 6-8 years and 75% in girls <6 years) and a diagnosis of exclusion.[23] However, an underlying cause must be excluded and actively searched for in very young female patients. In males, an underlying cause must be sought, as only around 60% of cases are classed as idiopathic.[24][25][26]
Tumors that can result in CPP include optic and hypothalamic gliomas, astrocytomas, ependymomas, and pineal tumors, and, rarely, primitive endocrine tumors such as a craniopharyngioma. Hamartomas of the tuber cinereum are congenital tumors composed of a heterotopic mass of gonadotropin-releasing hormone (GnRH) neurosecretory neurons, fiber bundles, and glial cells, that are frequently associated with CPP, often before 3 years of age.[46] They are often associated with gelastic epilepsy presenting often as giggling episodes and developmental delay.
Hydrocephalus, head injury, previous infections such as meningitis or encephalitis, and neurofibromatosis are also associated with CPP. A history of these must therefore be sought.
Past history may indicate surgery (due to a tumor) or radiation therapy to the brain. The prevalence of CPP is increased after cranial irradiation for local tumors or leukemia. Moderate radiation doses used for the treatment of brain tumors in children, as opposed to low or high doses, are associated with precocious puberty, with a direct relationship between the age at pubertal onset and therapy.[29] Higher doses are usually associated with gonadotropin deficiency and a delay in puberty.
Disconsonant or gonadotropin-independent precocious puberty (GIPP)
Disconsonant pubertal development suggests a peripheral cause, meaning that secretion of sex steroids is autonomous and independent of the hypothalamic GnRH pulse generator. Boys present with greater virilization (penile and scrotal development) but inappropriately small testes. Girls may have advanced pubic and axillary hair growth prior to breast development.
There may be a history of congenital adrenal hyperplasia (CAH) since birth or it may be a late diagnosis. The child may already be on hydrocortisone and/or fludrocortisone therapy. Males with 21-hydroxylase CAH can present with GIPP if undertreated for prolonged periods. Females with 21-hydroxylase CAH present with signs of virilization (e.g., pubic and axillary hair, clitoromegaly) due to excess androgen but no breast development.
Testotoxicosis (also called male-limited GIPP) is associated with a number of constitutively activating mutations of the luteinizing hormone (LH) receptor.[43][44] It is also associated with premature Leydig cell and germ cell maturation. The disorder is inherited as an autosomal dominant condition that manifests only in males. Family history may help identify affected patients.
There may be a history of bone deformity or other endocrine gland hyperfunctioning due to McCune-Albright syndrome (MAS). This is a multisystem disorder, characterized by the classic triad of GIPP with irregularly edged hyperpigmented macules or café au lait spots and a slowly progressive bone disorder (polyostotic fibrous dysplasia).[47][48][49] Symptoms of other endocrine involvement include thyrotoxicosis (thyroid nodular hyperplasia), Cushing syndrome (multiple adrenal hyperplastic nodules), gigantism or acromegaly (pituitary adenoma and growth hormone excess), and galactorrhea (due to hyperprolactinemia). Sexual precocity is rare in boys with MAS.
Prolonged sex-steroid exposure in GIPP has a direct maturational effect on the hypothalamus and can accelerate the onset of centrally mediated or GnRH-dependent puberty (CPP).
Examination
Present and previous height and weight measurements should be accurately recorded. Accurate measurements, using correct age- and sex-specific growth charts, are mandatory. Care must be taken to plot the height and weight based on the child's actual chronologic age. Growth is strongly related to the genetic potential. The target or midparental height is calculated as follows:
Girl = ([height of mother + height of father]/2) - 7 cm (2.5 inches)
Boy = ([height of mother + height of father]/2) + 7 cm (2.5 inches)
Growth velocity determines the change in height over time. It is calculated as the difference in height on two different occasions annualized over a year. A height that plots consistently along a given percentile on the growth chart reflects normal growth velocity. Crossing percentiles in an upward direction reflects an accelerated growth velocity.
Patients with CPP have a pubertal examination consonant with normal pubertal development. In GIPP in boys, virilization occurs due to high concentrations of testosterone. However, enlargement of the testes is absent or only present in the early to mid-pubertal range, and is smaller than expected in relation to the stage of penile growth. The testes are modestly and symmetrically enlarged in testotoxicosis but asymmetrical with Leydig cell tumors. In females, there may be little or no breast development despite the presence of pubic or axillary hair and/or menses.
Presence of dysmorphic features may reveal multisystem syndromes such as midline facial abnormalities in holoprosencephaly and septo-optic dysplasia. Facial asymmetry may be observed in MAS.
Skin examination should help identify café au lait spots present in neurofibromatosis (regular edges) and MAS (irregular edges). There may also be other skin involvement in neurofibromatosis type 1 (NF1), such as plexiform neurofibromas or intertriginous or axillary freckling.
A detailed examination of the eyes should always be performed, including visual field assessment and fundoscopy. Visual abnormalities include Lisch nodules in neurofibromatosis type 1, or other visual deficits due to brain injury, tumors, or infections, or optic nerve hypoplasia in septo-optic dysplasia. Focal motor deficits may be present due to intracranial tumors, previous head injury, hydrocephalus, meningitis, or encephalitis. [Figure caption and citation for the preceding image starts]: Male with consonant pubertal developmentFrom the collection of Dr A. Mehta; used with permission [Citation ends].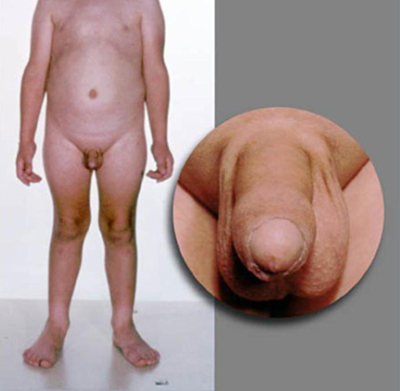 [Figure caption and citation for the preceding image starts]: Male with congenital adrenal hyperplasia and disconsonant pubertal developmentFrom the collection of Dr A. Mehta; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Male with congenital adrenal hyperplasia and disconsonant pubertal developmentFrom the collection of Dr A. Mehta; used with permission [Citation ends].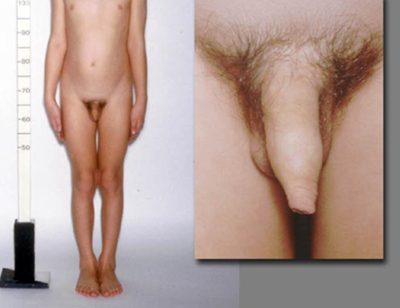 [Figure caption and citation for the preceding image starts]: Female with GIPP and café au lait hyperpigmented macules in McCune-Albright SyndromeFrom the collection of Dr A. Mehta; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Female with GIPP and café au lait hyperpigmented macules in McCune-Albright SyndromeFrom the collection of Dr A. Mehta; used with permission [Citation ends]. [Figure caption and citation for the preceding image starts]: Axillary freckling in neurofibromatosis type IFrom the collection of Dr A. Mehta; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Axillary freckling in neurofibromatosis type IFrom the collection of Dr A. Mehta; used with permission [Citation ends].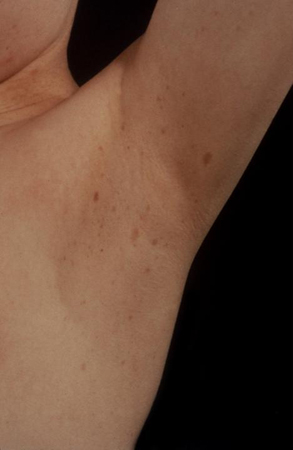
Initial investigations
Idiopathic precocious puberty accounts for the majority of cases in girls, but few cases in boys, and is a diagnosis of exclusion. Initial investigations should include the following elements.
A nondominant (typically, left) hand/wrist radiograph to estimate skeletal age. The appearance of representative epiphyseal centers on the x-ray is compared with age and sex-appropriate published standards. The most commonly used method is that of Greulich and Pyle or an automated bone age report such as BoneXpert.[50] The bone age is typically advanced by 1-3 years in patients with precocious puberty. The bone age may also help predict the estimated adult height range and its relation to the midparental height; caution must be exercised in this interpretation as current standards have been validated in normal children only and not in those with precocious puberty.
The biochemical diagnosis of CPP is based on pubertal serum gonadotropin concentrations either as basal levels or after stimulation. Before the onset of puberty, follicle-stimulating hormone (FSH) is usually the predominant gonadotropin, while during and after puberty luteinizing hormone (LH) concentrations are higher than FSH. Basal LH is much more sensitive than basal FSH concentration. In cases of CPP, basal LH concentration is usually ≥0.3 IU/L. An elevated basal LH has a high sensitivity and specificity for the diagnosis of precocious puberty in males, but is less sensitive in females. Thus, in girls, basal LH measurement may be adequate to confirm, but not to refute, the diagnosis of CPP.[40]
The GnRH test is used in less clearcut cases. This involves measuring LH and FSH levels at baseline, and then again at 20 and 60 minutes after administration of GnRH. A pubertal response is considered with a serum LH concentration after stimulation of ≥5 IU/L.[51] GnRH analogs have also been used for the investigation of peak LH and FSH concentrations following stimulation if recombinant GnRH is unavailable.[52]
Elevated serum estrogen and testosterone measurement in girls and boys respectively help confirm the onset of puberty.
A pelvic ultrasound scan can help in the diagnosis of precocious puberty in girls. The uterus may have a pre, peri, or postpubertal shape and size depending on the stage of puberty. An endometrial echo reflects an estrogen effect, while the presence of follicles in the ovaries reflects gonadotropin stimulation. Endometrial thickening suggests that pubertal concentrations of estrogen have been attained, and an endometrium of around 6-8 mm implies imminent menarche. The sexual precocity in girls with MAS is caused by autonomously functioning multiple, luteinized, follicular cysts of the ovaries with an occasional large solitary cyst. A pelvic ultrasound will also help identify gonadal estrogen-secreting tumors.
If the suspected diagnosis is premature adrenarche (which is typically associated with pubic hair and/or body odor) then LH, FSH, estradiol, or testosterone may not be necessary.[53]
Subsequent tests
Further investigations and management should depend on the symptoms, pattern of sexual development, sex, and results of gonadotropin measurement.
CPP
Magnetic resonance imaging (MRI) of the brain helps identify tumors, presence of hydrocephalus, structural hypothalamo-pituitary abnormalities, and midline brain defects. The characteristic appearance of a hamartoma is that of a sessile or pedunculated mass usually attached to the posterior hypothalamus between the tuber cinereum and the mamillary bodies.[54] There is consensus that MRI of the brain needs to be done in all boys and MRI of the brain in girls with onset of puberty prior to age 6, but studies have found that the risk of finding a tumor or another serious lesion is very low (<1%) in girls with onset between ages 6 and 8 years, so many advise not routinely imaging such girls.[55]
Presence of CPP in certain situations such as post-surgery for a craniopharyngioma, following radiation therapy, or in patients with septo-optic dysplasia and holoprosencephaly, should warrant a full pituitary hormone evaluation to look for abnormalities (most commonly deficiencies) of other anterior and posterior pituitary hormones.
Overnight sampling of gonadotropins may demonstrate pulsatility. It is primarily undertaken in a research setting.
GIPP
Serum 17-hydroxyprogesterone (17-OHP) and other androgens (androstenedione, dehydroepiandrostenedione sulfate) to diagnose 21-hydroxylase or other types of CAH. A urinary steroid profile and adrenocorticotropin stimulation test may help identify adrenal steroid synthesis defects.
If an adrenal tumor is suspected in patients with virilization, a computed tomographic scan (or MRI) of the adrenal glands is indicated. Ultrasound scan of the adrenal glands is of limited use.
In McCune-Albright syndrome, depending on symptomatology, other investigations may include an MRI of the adrenals for multiple adrenal hyperplastic nodules causing Cushing syndrome, thyroid scan for nodular hyperplasia presenting with thyrotoxicosis, or MRI brain in the presence of gigantism or galactorrhea to rule out a pituitary adenoma. A bone scan and a skeletal survey to identify the bony lesions of polyostotic fibrous dysplasia are also indicated.
Other tests to be considered:
Genetic testing should be carried out in patients suspected to have neurofibromatosis type 1, MAS, or familial testotoxicosis. Loss-of-function mutations in MKRN3 or DLK1, or gain-of-function mutations in KISS1R, a G protein-coupled receptor that is a ligand for kisspeptin, can cause CPP.[31]
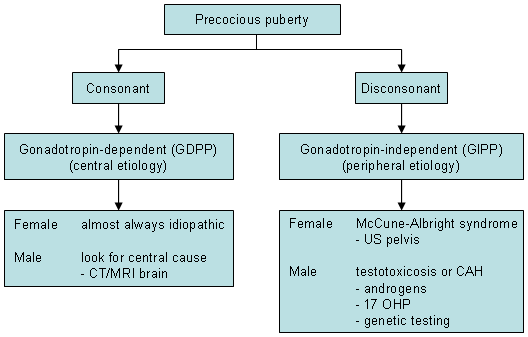
Thyroid function tests to rule out primary hypothyroidism, a rare cause of early puberty. This is mainly helpful in cases where symptoms are suggestive of hypothyroidism and where growth velocity is decreased instead of increased. [Figure caption and citation for the preceding image starts]: Investigations in patients with precocious pubertyFrom the collection of Dr A. Mehta; used with permission [Citation ends].
Use of this content is subject to our disclaimer