Diagnosis of Lambert-Eaton myasthenic syndrome (LEMS) is supported by electrophysiologic and serologic tests. Cancer is found either at disease onset or subsequently in 40% to 54% of patients with cancer-associated LEMS (CA-LEMS); small cell lung cancer (SCLC) is the most commonly associated cancer.[2]O'Neill JH, Murray NM, Newsom-Davis J. The Lambert-Eaton myasthenic syndrome. A review of 50 cases. Brain. 1988 Jun;111(pt 3):577-96.
http://www.ncbi.nlm.nih.gov/pubmed/2838124?tool=bestpractice.com
[8]Titulaer MJ, Wirtz PW, Willems LN, et al. Screening for small-cell lung cancer: a follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol. 2008 Sep 10;26(26):4276-81.
http://www.ncbi.nlm.nih.gov/pubmed/18779614?tool=bestpractice.com
[9]Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 2011 Dec;10(12):1098-107.
http://www.ncbi.nlm.nih.gov/pubmed/22094130?tool=bestpractice.com
As such, it is essential to thoroughly evaluate for an underlying cancer.
In the absence of malignancy, demonstrating coexisting autoimmune disease early in the course of LEMS may increase the probability of non-cancer-associated LEMS (NCA-LEMS). Thyroid-stimulating hormone should be measured in all patients with LEMS to assess for comorbid thyroid dysfunction. Assessment for other coexisting autoimmune disease (e.g., rheumatoid arthritis, systemic lupus erythematosus [SLE], systemic vasculitis, pernicious anemia) should be considered if the patient is symptomatic. However, routine surveillance for autoimmune disorders should not be necessary unless prompted by symptoms.
History and physical exam
History should include an assessment of known risk factors for SCLC (e.g., history of cigarette smoking), and for any coexisting autoimmune disorder or family history of autoimmune disease.[10]Wirtz PW, Bradshaw J, Wintzen AR, et al. Associated autoimmune diseases in patients with the Lambert-Eaton myasthenic syndrome and their families. J Neurol. 2004 Oct;251(10):1255-9.
http://www.ncbi.nlm.nih.gov/pubmed/15503107?tool=bestpractice.com
[12]Wirtz PW, Smallegange TM, Wintzen AR, et al. Differences in clinical features between the Lambert-Eaton myasthenic syndrome with and without cancer: an analysis of 227 published cases. Clin Neurol Neurosurg. 2002 Sep;104(4):359-63.
http://www.ncbi.nlm.nih.gov/pubmed/12140105?tool=bestpractice.com
In LEMS, the age of symptom onset is later in life, typically in the 50s and 60s, although children and young adults may rarely be affected. Symptom onset is usually insidious but may be more acute in association with an infection, with exposure to neuromuscular blocking agents, or in association with malignancy. Dyspnea may indicate severe respiratory or bulbar weakness and is a neurologic emergency.
Generalized fatigue, proximal leg weakness, and dry mouth are common initial symptoms and may fluctuate throughout the day. Weakness begins in the proximal legs, typically hip flexion and hip abduction, with variable progression, and subsequently usually affects the proximal arms. Distal limb muscles may be involved; this is more common in CA-LEMS.[1]Titulaer MJ, Wirtz PW, Kuks JB, et al. The Lambert-Eaton myasthenic syndrome 1988-2008: a clinical picture in 97 patients. J Neuroimmunol. 2008 Sep 15;201-2:153-8.
http://www.ncbi.nlm.nih.gov/pubmed/18644631?tool=bestpractice.com
Objective weakness is usually mild compared with reported symptoms. Tendon reflexes are reduced or absent in most patients, but may be preserved early in the disease. Hypoactive reflexes can often be potentiated by brief, isometric contraction of the appropriate muscle.[2]O'Neill JH, Murray NM, Newsom-Davis J. The Lambert-Eaton myasthenic syndrome. A review of 50 cases. Brain. 1988 Jun;111(pt 3):577-96.
http://www.ncbi.nlm.nih.gov/pubmed/2838124?tool=bestpractice.com
Strength may improve after exercise and then weaken as activity is sustained. A "waddling" gait may be noted.
Dysarthria is common, occurring in up to 71% of patients with LEMS, although it typically occurs late in the disease course.[1]Titulaer MJ, Wirtz PW, Kuks JB, et al. The Lambert-Eaton myasthenic syndrome 1988-2008: a clinical picture in 97 patients. J Neuroimmunol. 2008 Sep 15;201-2:153-8.
http://www.ncbi.nlm.nih.gov/pubmed/18644631?tool=bestpractice.com
The presence of prominent ocular weakness with ptosis and binocular diplopia early in the course may simulate myasthenia gravis. Dysphagia is reported in up to 43% of patients with LEMS. This is important to recognize, because prominent swallowing difficulty may necessitate more aggressive therapy.[1]Titulaer MJ, Wirtz PW, Kuks JB, et al. The Lambert-Eaton myasthenic syndrome 1988-2008: a clinical picture in 97 patients. J Neuroimmunol. 2008 Sep 15;201-2:153-8.
http://www.ncbi.nlm.nih.gov/pubmed/18644631?tool=bestpractice.com
Dry mouth (xerostomia) is the most frequently encountered autonomic symptom, occurring in up to 78% of patients, and often precedes other symptoms.[1]Titulaer MJ, Wirtz PW, Kuks JB, et al. The Lambert-Eaton myasthenic syndrome 1988-2008: a clinical picture in 97 patients. J Neuroimmunol. 2008 Sep 15;201-2:153-8.
http://www.ncbi.nlm.nih.gov/pubmed/18644631?tool=bestpractice.com
This may be experienced by the patient as a "metallic taste." Other manifestations include pupillary dilation or orthostatic hypotension. Male impotence as well as progression of weakness to extraocular, bulbar, and distal muscles within 6 months of onset is highly suggestive of CA-LEMS.[1]Titulaer MJ, Wirtz PW, Kuks JB, et al. The Lambert-Eaton myasthenic syndrome 1988-2008: a clinical picture in 97 patients. J Neuroimmunol. 2008 Sep 15;201-2:153-8.
http://www.ncbi.nlm.nih.gov/pubmed/18644631?tool=bestpractice.com
Evidence of cerebellar ataxia is uncommon; if present, it suggests a more widespread paraneoplastic syndrome associated with an underlying SCLC with a paraneoplastic cerebellar syndrome with reduced P/Q-type voltage-gated calcium-channels (VGCCs) in the molecular layer of the cerebellum.[1]Titulaer MJ, Wirtz PW, Kuks JB, et al. The Lambert-Eaton myasthenic syndrome 1988-2008: a clinical picture in 97 patients. J Neuroimmunol. 2008 Sep 15;201-2:153-8.
http://www.ncbi.nlm.nih.gov/pubmed/18644631?tool=bestpractice.com
[21]Fukuda T, Motomura M, Nakao Y, et al. Reduction of P/Q-type calcium channels in the postmortem cerebellum of paraneoplastic cerebellar degeneration with Lambert-Eaton myasthenic syndrome. Ann Neurol. 2003 Jan;53(1):21-8.
http://www.ncbi.nlm.nih.gov/pubmed/12509844?tool=bestpractice.com
Electrophysiology
Electrophysiologic testing should be performed for most patients. Nerve conduction studies and low-frequency repetitive nerve stimulation (RNS) are performed as initial tests.
Nerve conduction studies: the initial compound muscle action potential amplitude is typically low in LEMS. After 10 seconds of isometric exercise, facilitation of ≥100% is found in at least one muscle in 90% of patients with LEMS. Postexercise facilitation varies but is greater in distal muscles.[22]Tim RW, Massey JM, Sanders DB. Lambert-Eaton myasthenic syndrome (LEMS). Clinical and electrodiagnostic features and response to therapy in 59 patients. Ann N Y Acad Sci. 1998 May 13;841:823-6.
http://www.ncbi.nlm.nih.gov/pubmed/9668336?tool=bestpractice.com
VGCC antibody-negative patients may have less pronounced facilitation.[23]Oh SJ, Hatanaka Y, Claussen GC, et al. Electrophysiological differences in seropositive and seronegative Lambert-Eaton myasthenic syndrome. Muscle Nerve. 2007 Feb;35(2):178-83.
http://www.ncbi.nlm.nih.gov/pubmed/17058271?tool=bestpractice.com
Low-frequency RNS: >10% decrement in amplitude between the first and fourth compound muscle action potential elicited by low-frequency stimulation (2-3 Hz) is a sensitive measure for LEMS, with nearly 100% sensitivity when distal limb muscles are studied.[22]Tim RW, Massey JM, Sanders DB. Lambert-Eaton myasthenic syndrome (LEMS). Clinical and electrodiagnostic features and response to therapy in 59 patients. Ann N Y Acad Sci. 1998 May 13;841:823-6.
http://www.ncbi.nlm.nih.gov/pubmed/9668336?tool=bestpractice.com
False-negatives may be seen in cool limbs and/or with incompletely relaxed muscles. This finding is relatively less specific, because it may be observed in other neuromuscular junction diseases (e.g., myasthenia gravis) and motor neuropathic processes (e.g., amyotrophic lateral sclerosis). A pattern of progressive compound muscle action potential amplitude decrement is typically observed in LEMS instead of the classic “saddle-shaped” pattern seen in myasthenia gravis.[24]Sanders DB, Cao L, Massey JM, et al. Is the decremental pattern in Lambert-Eaton syndrome different from that in myasthenia gravis? Clin Neurophysiol. 2014 Jun;125(6):1274-7.
http://www.ncbi.nlm.nih.gov/pubmed/24332471?tool=bestpractice.com
[25]Baslo MB, Deymeer F, Serdaroglu P, et al. Decrement pattern in Lambert-Eaton myasthenic syndrome is different from myasthenia gravis. Neuromuscul Disord. 2006 Jul;16(7):454-8.
http://www.ncbi.nlm.nih.gov/pubmed/16806929?tool=bestpractice.com
High-frequency RNS: RNS performed at 20-50 Hz to demonstrate tetanic facilitation has no significant diagnostic superiority to eliciting postexercise facilitation with 10 seconds of maximum voluntary isometric exercise. At least 100% or greater postexercise or tetanic facilitation with high-frequency RNS in the abductor digiti quinti manus muscle is fairly specific for the diagnosis of LEMS.[23]Oh SJ, Hatanaka Y, Claussen GC, et al. Electrophysiological differences in seropositive and seronegative Lambert-Eaton myasthenic syndrome. Muscle Nerve. 2007 Feb;35(2):178-83.
http://www.ncbi.nlm.nih.gov/pubmed/17058271?tool=bestpractice.com
[26]AAEM Quality Assurance Committee, American Association of Electrodiagnostic Medicine. Practice parameter for repetitive nerve stimulation and single fiber EMG evaluation of adults with suspected myasthenia gravis or Lambert-Eaton myasthenic syndrome: summary statement. Muscle Nerve. 2001 Sep;24(9):1236-8.
http://www.aanem.org/AANEM/media/AANEM/Documents/Practice/Practice%20Guidelines/myastheniagravis.pdf
http://www.ncbi.nlm.nih.gov/pubmed/11494280?tool=bestpractice.com
In LEMS, the postexercise facilitation lasts a few seconds, as opposed to botulism where facilitation may be sustained for several minutes.
Single-fiber EMG: a highly sensitive investigation for neuromuscular junction dysfunction.[26]AAEM Quality Assurance Committee, American Association of Electrodiagnostic Medicine. Practice parameter for repetitive nerve stimulation and single fiber EMG evaluation of adults with suspected myasthenia gravis or Lambert-Eaton myasthenic syndrome: summary statement. Muscle Nerve. 2001 Sep;24(9):1236-8.
http://www.aanem.org/AANEM/media/AANEM/Documents/Practice/Practice%20Guidelines/myastheniagravis.pdf
http://www.ncbi.nlm.nih.gov/pubmed/11494280?tool=bestpractice.com
[27]Oh SJ, Ohira M. Single-fiber EMG and clinical correlation in Lambert-Eaton myasthenic syndrome. Muscle Nerve. 2001 Sep;24(9):1236-8.
http://www.ncbi.nlm.nih.gov/pubmed/23505075?tool=bestpractice.com
Specialized needle electrode recordings from ≥2 muscle fibers innervated by the same motor axon exhibit abnormally increased variability in time intervals between muscle fiber action potentials (jitter), or failure of neuromuscular transmission (blocking). As in other presynaptic neuromuscular junction disorders (e.g., botulism), jitter and blocking in LEMS are rate-dependent and improve with higher rates of motor axonal firing, either with voluntary activation or with axonal stimulation.[Figure caption and citation for the preceding image starts]: Compound muscle action potentials (abductor digiti quinti manus muscle) following ulnar nerve stimulation: (A) at rest, (B) immediately after 10 seconds of maximum voluntary contraction demonstrating 1500% postexercise facilitationFrom the collection of Dr Vern C. Juel [Citation ends].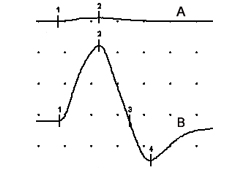
Autoantibody serology
Serology for VGCCs should be assessed. VGCC antibodies are reported in 76% to 95% of patients with LEMS.[3]Sanders DB, Juel VC. Chapter 9 The Lambert-Eaton myasthenic syndrome. Handb Clin Neurol. 2008;91:273-83.
http://www.ncbi.nlm.nih.gov/pubmed/18631847?tool=bestpractice.com
[20]Lennon VA, Kryzer TJ, Griesmann GE, et al. Calcium-channel antibodies in the Lambert-Eaton syndrome and other paraneoplastic syndromes. N Engl J Med. 1995 Jun 1;332(22):1467-74.
http://www.nejm.org/doi/full/10.1056/NEJM199506013322203#t=article
http://www.ncbi.nlm.nih.gov/pubmed/7739683?tool=bestpractice.com
[28]Nakao YK, Motomura M, Fukudome T, et al. Seronegative Lambert-Eaton myasthenic syndrome: study of 110 Japanese patients. Neurology. 2002 Dec 10;59(11):1773-5.
http://www.ncbi.nlm.nih.gov/pubmed/12473768?tool=bestpractice.com
However, antibody titers do not correlate with disease severity, and titers may fall or disappear with immunosuppression.[29]Leys K, Lang B, Johnston I, et al. Calcium channel autoantibodies in the Lambert-Eaton myasthenic syndrome. Ann Neurol. 1991 Mar;29(3):307-14.
http://www.ncbi.nlm.nih.gov/pubmed/1645944?tool=bestpractice.com
Furthermore, low VGCC antibody titers have been found in systemic lupus erythematosus and rheumatoid arthritis, and in some patients (<5%) with myasthenia gravis, though the omega-conotoxin assay may provide additional specificity.[20]Lennon VA, Kryzer TJ, Griesmann GE, et al. Calcium-channel antibodies in the Lambert-Eaton syndrome and other paraneoplastic syndromes. N Engl J Med. 1995 Jun 1;332(22):1467-74.
http://www.nejm.org/doi/full/10.1056/NEJM199506013322203#t=article
http://www.ncbi.nlm.nih.gov/pubmed/7739683?tool=bestpractice.com
[30]Lang B, Johnston I, Leys K, et al. Autoantibody specificities in Lambert-Eaton myasthenic syndrome. Ann N Y Acad Sci. 1993 Jun 21;681:382-93.
http://www.ncbi.nlm.nih.gov/pubmed/8395152?tool=bestpractice.com
Additional autoantibody serology may also be of benefit.
The presence of antibodies in response to domain IV of the alpha-1A P/Q VGCC subunit is highly suggestive of non-cancer-associated LEMS (38% of patients with NCA-LEMS compared with 5% of patients with CA-LEMS).[31]Pellkofer HL, Armbruster L, Krumbholz M, et al. Lambert-Eaton myasthenic syndrome differential reactivity of tumor versus non-tumor patients to subunits of the voltage-gated calcium channel. J Neuroimmunol. 2008 Nov 15;204(1-2):136-9.
http://www.ncbi.nlm.nih.gov/pubmed/18809213?tool=bestpractice.com
The presence of SOX1 antibodies shows 64% sensitivity in CA-LEMS for underlying SCLC. SOX1 antibodies are rarely seen in NCA-LEMS; therefore, the presence of SOX1 antibodies should prompt a thorough search for SCLC. However, these antibodies are also seen in Hu-positive paraneoplastic neurologic syndromes (32%) and in SCLC without neurologic symptoms (22%).[32]Sabater L, Titulaer M, Saiz A, et al. SOX1 antibodies are markers of paraneoplastic Lambert-Eaton myasthenic syndrome. Neurology. 2008 Mar 18;70(12):924-8.
http://www.ncbi.nlm.nih.gov/pubmed/18032743?tool=bestpractice.com
The presence of acetylcholine receptor (AChR) or MuSK antibodies strongly suggests myasthenia gravis, although up to 13% of patients with LEMS have AChR antibodies, and myasthenia gravis/LEMS overlap syndromes do occur rarely.[33]Lennon VA. Serologic profile of myasthenia gravis and distinction from the Lambert-Eaton myasthenic syndrome. Neurology. 1997;48(suppl 5):S23-S27.
HLA typing
Associated HLA haplotypes may also be assessed. The presence of specific haplotypes is not diagnostic for LEMS but may be of some value in distinguishing non-cancer-associated LEMS from cancer-associated LEMS. A higher frequency of HLA-DR3, -B8, or -A1 is seen in NCA-LEMS (HLA-DR3, 67%; -B8, 64%; -A1, 52%) than in CA-LEMS (HLA-DR, 30%; -B8, 20%; -A1, 18%).[7]Titulaer MJ, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: tumor versus nontumor forms. Ann N Y Acad Sci. 2008;1132:129-34.
http://www.ncbi.nlm.nih.gov/pubmed/18567862?tool=bestpractice.com
Forty-one percent of patients with non-cancer-associated LEMS have all three haplotypes compared with 5% of patients with cancer-associated LEMS.[7]Titulaer MJ, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: tumor versus nontumor forms. Ann N Y Acad Sci. 2008;1132:129-34.
http://www.ncbi.nlm.nih.gov/pubmed/18567862?tool=bestpractice.com
Other studies
A chest CT scan should be ordered for all patients with suspected CA-LEMS.[34]Ivanovski T, Miralles F. Lambert-Eaton myasthenic syndrome: early diagnosis is key. Degener Neurol Neuromuscul Dis. 2019 May 13:9:27-37.
https://www.dovepress.com/lambert-eaton-myasthenic-syndrome-early-diagnosis-is-key-peer-reviewed-fulltext-article-DNND
http://www.ncbi.nlm.nih.gov/pubmed/31191084?tool=bestpractice.com
Cancer is found either at disease onset or subsequently in around 50% of patients with LEMS.[2]O'Neill JH, Murray NM, Newsom-Davis J. The Lambert-Eaton myasthenic syndrome. A review of 50 cases. Brain. 1988 Jun;111(pt 3):577-96.
http://www.ncbi.nlm.nih.gov/pubmed/2838124?tool=bestpractice.com
[8]Titulaer MJ, Wirtz PW, Willems LN, et al. Screening for small-cell lung cancer: a follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol. 2008 Sep 10;26(26):4276-81.
http://www.ncbi.nlm.nih.gov/pubmed/18779614?tool=bestpractice.com
[9]Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 2011 Dec;10(12):1098-107.
http://www.ncbi.nlm.nih.gov/pubmed/22094130?tool=bestpractice.com
SCLC is the most commonly associated cancer; LEMS precedes the diagnosis of SCLC in up to 69% of patients with LEMS.[3]Sanders DB, Juel VC. Chapter 9 The Lambert-Eaton myasthenic syndrome. Handb Clin Neurol. 2008;91:273-83.
http://www.ncbi.nlm.nih.gov/pubmed/18631847?tool=bestpractice.com
[7]Titulaer MJ, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: tumor versus nontumor forms. Ann N Y Acad Sci. 2008;1132:129-34.
http://www.ncbi.nlm.nih.gov/pubmed/18567862?tool=bestpractice.com
If an initial chest CT is negative, follow-up CT scans and other imaging should be considered.[8]Titulaer MJ, Wirtz PW, Willems LN, et al. Screening for small-cell lung cancer: a follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol. 2008 Sep 10;26(26):4276-81.
http://www.ncbi.nlm.nih.gov/pubmed/18779614?tool=bestpractice.com
SCLC is subsequently diagnosed in up to 96% of patients with CA-LEMS within 1 year of diagnosis.[8]Titulaer MJ, Wirtz PW, Willems LN, et al. Screening for small-cell lung cancer: a follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol. 2008 Sep 10;26(26):4276-81.
http://www.ncbi.nlm.nih.gov/pubmed/18779614?tool=bestpractice.com
Total-body fluoro-2-deoxyglucose positron emission tomography may detect SCLC when chest CT is normal.[8]Titulaer MJ, Wirtz PW, Willems LN, et al. Screening for small-cell lung cancer: a follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol. 2008 Sep 10;26(26):4276-81.
http://www.ncbi.nlm.nih.gov/pubmed/18779614?tool=bestpractice.com
[34]Ivanovski T, Miralles F. Lambert-Eaton myasthenic syndrome: early diagnosis is key. Degener Neurol Neuromuscul Dis. 2019 May 13:9:27-37.
https://www.dovepress.com/lambert-eaton-myasthenic-syndrome-early-diagnosis-is-key-peer-reviewed-fulltext-article-DNND
http://www.ncbi.nlm.nih.gov/pubmed/31191084?tool=bestpractice.com
[35]Titulaer MJ, Soffietti R, Dalmau J, et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol. 2011 Jan;18(1):19-e3.
http://onlinelibrary.wiley.com/doi/10.1111/j.1468-1331.2010.03220.x/full
http://www.ncbi.nlm.nih.gov/pubmed/20880069?tool=bestpractice.com
If these studies are uninformative and there is significant risk of lung cancer, especially in smokers, or if symptoms have been present for <2 years, bronchoscopy may be of benefit, although one study suggests that the yield would be low.[3]Sanders DB, Juel VC. Chapter 9 The Lambert-Eaton myasthenic syndrome. Handb Clin Neurol. 2008;91:273-83.
http://www.ncbi.nlm.nih.gov/pubmed/18631847?tool=bestpractice.com
[8]Titulaer MJ, Wirtz PW, Willems LN, et al. Screening for small-cell lung cancer: a follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol. 2008 Sep 10;26(26):4276-81.
http://www.ncbi.nlm.nih.gov/pubmed/18779614?tool=bestpractice.com
Serial PFTs may be indicated in patients with shortness of breath and suspected respiratory crisis. Indication for mechanical ventilation includes FVC of ≤15 mL/kg and negative inspiratory force of ≤20 cm H₂O.
Neither abnormal ABGs nor pulse oxygenation reflects the degree of respiratory weakness because abnormalities in either occur late in the course after clinical decompensation.