Aetiology
Congenital GHD
Includes an expanding group of different aetiological disorders.[4][5] It may be isolated or occur with combined pituitary hormone deficiencies (CPHD) or with extra-pituitary features such as optic nerve hypoplasia, midline forebrain defects, intellectual disability, neck abnormalities, cerebellar abnormalities, or holoprosencephaly (syndromic hypopituitarism). Most cases are idiopathic in origin.
Perinatal complications such as prematurity, gestational bleeding, complicated delivery, and fetal distress or asphyxia may also be risk factors.[6] However, there is controversy over whether these are true risk factors or complications of hypopituitarism.
Congenital GHD may be sporadic or familial in 5% to 30% of cases. A rapidly expanding list of mutations in several pituitary transcription factors and genes have been identified for both GHD and CPHD (e.g., GHI, GHRHR, PROP1, POU1F1, HESX1, LHX3, LHX4, SOX3, OTX2, and RNPC3). Variable penetrance and oligogenic inheritance is common, and therefore interpretation of genetic findings requires specialist input.[7][8][9][10][11][12]
There are 6 defined types of familial isolated GHD (IGHD).
[Figure caption and citation for the preceding image starts]: Types of familial isolated growth hormone deficiency syndromes. CPHD, combined pituitary hormone deficiencies; GH, growth hormone; IGHD, isolated growth hormone deficiency; rhGH, recombinant human growth hormone; TSH, thyroid-stimulating hormone; PRL, prolactinCreated by the BMJ Knowledge Centre based on references cited in the topic [Citation ends].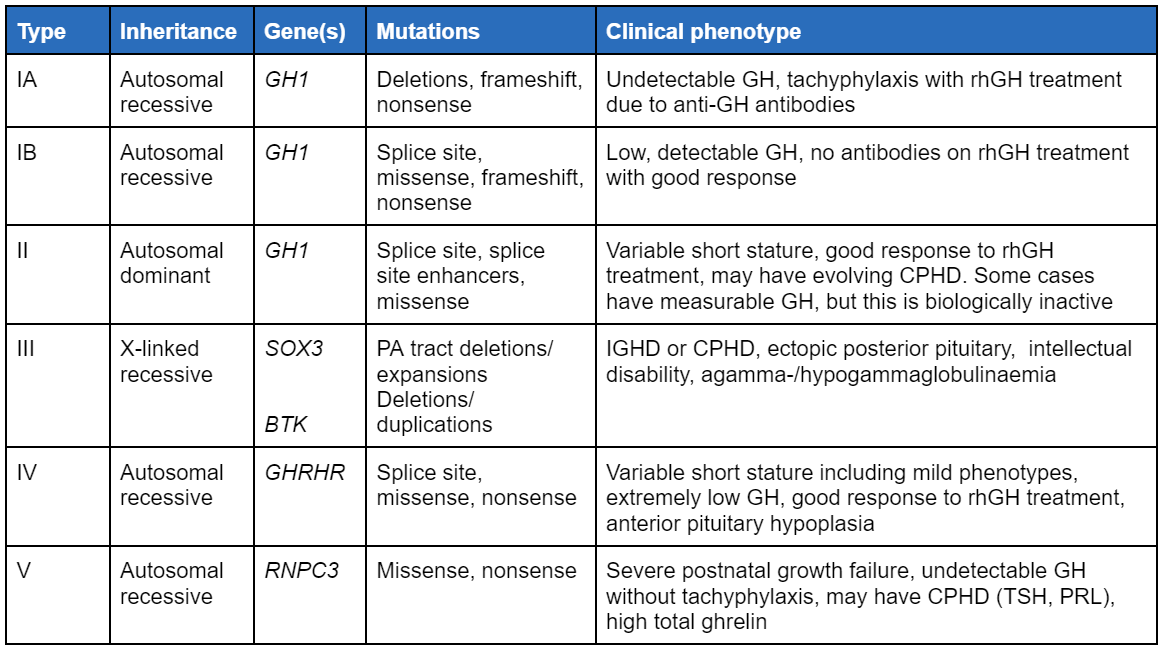
Acquired GHD
Hypothalamo-pituitary tumours: these can be benign or malignant, cystic or solid. GHD may arise due to the tumour, as a result of surgery, or after radiotherapy. The most common supra-/intrasellar tumour in childhood is a craniopharyngioma.[13][14][15] Other rare tumours seen in this region include pituitary adenomas, germinomas, optic pathway gliomas, hamartomas, meningiomas, and chordomas. Cystic lesions such as Rathke's cleft cysts, arachnoid cysts, or dermoid cysts, and, even more rarely, metastatic tumours such as childhood Hodgkin's and nasopharyngeal carcinomas, may also cause GHD.
Radiotherapy for central nervous system (CNS) tumours: exposure of the hypothalamo-pituitary region to any irradiation (locally and distally; e.g., medulloblastomas, ependymoma, or for haematological malignancies and bone marrow transplant) can result in profound GHD.[16]
Traumatic brain injury: pituitary damage may occur after traumatic brain injury (e.g., road traffic accidents, non-accidental injury) or after neurosurgery.[17][18]
Other rare causes include infections (e.g., meningitis, encephalitis, pituitary abscess); inflammatory/infiltrative disorders (e.g., sarcoidosis, tuberculosis, autoimmune lymphocytic hypophysitis, Langerhans cell histiocytosis); iron overload disorders (e.g., primary haemochromatosis, thalassaemia, and other diseases requiring chronic transfusions).[19]
A reversible GHD may also result after psychosocial deprivation.[20]
Pathophysiology
The pituitary gland includes the adenohypophysis (anterior and intermediate lobes) and neurohypophysis (posterior lobe). The anterior pituitary consists of somatotrophs producing GH, thyrotrophs secreting thyrotrophin or thyroid-stimulating hormone (TSH), corticotrophs secreting corticotrophin or adrenocorticotrophic hormone (ACTH), gonadotrophs producing follicle-stimulating hormone (FSH) and luteinising hormone (LH), and lactotrophs secreting prolactin.
Hypothalamic GH-releasing hormone (GHRH) stimulates the release of GH, and somatostatin inhibits GH release. GH release is pulsatile and occurs particularly during slow wave sleep, with higher amplitude pulses released in childhood than in adulthood. The role of endogenous ghrelin in normal growth is not known.[21][22] The human GH gene (GH) forms part of a cluster of 5 homologous genes and is located on the long arm of chromosome 17 (17q22-24).
GH secreted from pituitary somatotrophs binds to its receptors and activates a signalling cascade that eventually leads to the generation of insulin-like growth factor 1 (IGF1), which then mediates the various growth-promoting actions of GH. Concentrations of IGF1 correlate well with those of GH. However, low IGF1 levels may be observed in hypothyroidism, malnutrition, poorly controlled diabetes mellitus, and chronic diseases. Untreated children with GHD will be short and have delayed puberty, decreased pubertal growth spurt, and a final height standard deviation score (SDS) of -4 to -6.[21][22][23] GH also increases calcium absorption and bone density, is lipolytic and reduces body fat, and increases muscle mass.
Some data suggest that children with isolated GHD may have cognitive impairment, including lower IQ, Verbal Comprehension Index, Processing Speed Index, and Movement Assessment Battery for Children scores.[24] However, further studies are required to investigate the effects and impact of GHD on brain structure, cognitive function, and motor performance.
Somatotrophs constitute 40% to 50% of the pituitary gland cell population and are especially vulnerable to pressure effects and radiation injury. The rich vascular network of the hypothalamus and pituitary and the structure of the pituitary stalk make it vulnerable to the effects of trauma. As a result, GHD and growth failure are early endocrine manifestations, followed by gonadotrophin and TSH deficiency, after any insult to the hypothalamo-pituitary region. Tumours and other lesions in the hypothalamo-pituitary area may cause endocrine disturbance either directly or secondary to treatment (surgery, radiotherapy).
Classification
Aetiological classification
No formal classification exists, apart from the classification for isolated GHD (IGHD), which is based on genotype. GHD may be present since birth (although may be evident only in infancy or childhood) or acquired. It may also be classified as IGHD or combined pituitary hormone deficiencies (CPHD). Congenital IGHD may be sporadic or familial; 5% to 30% are familial and there are 6 defined types (see table in Aetiology section).
Use of this content is subject to our disclaimer