Approach
Infants with SCID typically present with recurrent and unusually severe infections (including opportunistic infections), faltering growth, and/or chronic diarrhoea. Key findings on examination may include the absence of lymphoid tissue, poor weight gain, or diffuse erythematous rash. Early diagnostic features include lymphopenia and the absence of a thymic shadow on chest x-ray. Specific diagnosis requires lymphocyte phenotyping (using flow cytometry) and molecular genetic testing.[22] Early diagnosis is critical as it leads to prompt treatment and reduced morbidity and mortality.[8][10][23][24][25]
In countries such as the US, where newborn screening (NBS) for SCID is routine, an early diagnosis can be made before signs and symptoms of SCID develop.[26][27]
History
The inheritance pattern for SCID is either X-linked or autosomal recessive. It is therefore essential to take a detailed family history, enquiring about family members with SCID and deaths of family members in early infancy.[4] A family history of early infant deaths secondary to infection or failure to thrive may be consistent with an inherited primary immunodeficiency such as SCID. In countries that have implemented NBS for SCID, the results of this test should be obtained.
A history of failure to thrive, opportunistic infections (e.g., Pneumocystis jirovecii), viral infections, recurrent pneumonias, thrush, or infections that do not resolve, despite appropriate antimicrobial treatment, warrant evaluation for SCID.[8][23] Due to placental transfer of maternal antibodies and isolation of newborns (common in some cultures), infections in infants with SCID may not occur in the first few months of life. With chronic infections, such as viral gastroenteritis, chronic diarrhoea is a common symptom.
History should include the HIV status of the mother and questioning to exclude secondary causes of immunodeficiency, such as medications (e.g., corticosteroids), metabolic diseases (e.g., diabetes mellitus, renal/hepatic failure), or infectious diseases (e.g., HIV, measles, varicella, cytomegalovirus [CMV], Epstein-Barr virus [EBV]). History of breastfeeding would be helpful to ascertain congenital or neonatal CMV infection.[28]
A family history of autoimmunity and malignancy should also be inquired about.[29]
Ethnic background and possibility of consanguinity should be assessed because there is an increased incidence of SCID in regions with high rates of consanguinity (e.g., Middle Eastern countries, Athabascan-speaking Native American people).[12][13][14][15][30]
Physical examination
Infants with SCID usually appear healthy at birth. Abnormalities on physical examination that may appear later in life include:[15][31]
Failure to thrive (i.e., decreased growth and poor weight gain)
Findings indicative of infection (e.g., tachypnoea, rales, oral thrush, and fungal infections of the skin or nails)
Absence of lymphoid tissue (tonsils and lymph nodes) may be noted, but can be difficult to detect in young infants.
Specific dysmorphic features can develop, for example:
Skeletal abnormalities, blindness, and dystonia in patients with adenosine deaminase deficiency SCID.[23]
Oral and genital ulcers in patients with Artemis/DNA cross-link repair 1C (DCLRE1C) SCID.[32]
Microcephaly in patients with DNA ligase IV-associated SCID.[33]
A diffuse erythrodermal rash in the first month of life in patients with hypomorphic variants of SCID.[34] A similar cutaneous presentation (along with hepatosplenomegaly and eosinophilia) can be seen in infants with SCID with maternal T-cell engraftment occurring in utero (i.e., a sign of graft-versus-host disease), and with non-irradiated blood products.[31] Presence of erythroderma or rash also suggests the possibility of a differential diagnosis of Omenn's syndrome, a clinical diagnosis associated with several SCID-associated genetic defects.[4]
Radiation sensitivity in patients with DCLRE1C SCID, DNA ligase IV deficiency, or Cernunnos/XLF deficiency.[35][36][37]
Laboratory investigations
Absolute lymphocyte count
A full blood count (FBC) with differential and calculation of the absolute lymphocyte count (ALC) is the initial laboratory investigation for all patients with suspected SCID. Lymphopenia on a routine FBC should also alert the physician to consider SCID.
ALC is calculated by multiplying the percentage of lymphocytes by the absolute white blood cell count. An ALC of <3000 cells/mm³ has been proposed as the cutoff to determine which infants require further evaluation for SCID.[38]
ALC may be normal in cases of maternal engraftment of lymphocytes, or in conditions where gene expression is partially intact (e.g., hypomorphic SCID variants), or in cases where B cells make up the majority of the lymphocyte population (e.g., T-B+ SCID).
Flow cytometry
Flow cytometry is required for diagnosing SCID and classifying patients according to lymphocyte phenotype. It is the initial diagnostic test for all patients with suspected SCID.[2][4]
Flow cytometry measures the total numbers of T cells, T-cell subsets (i.e., CD4+ T cells, CD8+ T cells), naive T cells (CD4+CD45RA+), memory T cells (CD4+CD45RO+), B cells, and natural killer (NK) cells in the peripheral blood.
The phenotypic classification of SCID is based on the absence or presence of B cells (B- or B+) and NK cells (NK- or NK+), in addition to the absence of T cells (T-).[2][3]
In healthy infants, the majority of T cells are naive (e.g., CD45RA isotype +). In all forms of SCID, there is a profound decrease in the number of all T cells including naive T cells. Maternal engraftment of T cells or residual gene function in hypomorphic variants of SCID may result in low to normal numbers of T cells, but these T cells are almost all memory T cells (CD4+CD45RO+).[8]
The Primary Immune Deficiency Treatment Consortium defines SCID as absent, or very low numbers of T cells (CD3 T cells <300/microlitre), and absent or very low T-cell function (<10% of lower limit of normal), as measured by T-cell proliferation studies using phytohaemagglutinin (PHA).[39]
Quantitative immunoglobulin test (IgG, IgM, and IgA)
Performed in all patients with suspected SCID to help confirm diagnosis.
All SCID patients (including those with normal B cell numbers) have hypogammaglobulinaemia secondary to the lack of T-cell help to induce antibody production.[4] Patients also have absent isohaemagglutinins and absent specific antibody responses to inactivated protein-based vaccines, such as tetanus and diphtheria.[34]
Newborn infants commonly have measurable levels of IgG due to placental transfer of maternal IgG to the fetus, but IgM and IgA are absent, or levels are low.
Imaging
A chest x-ray demonstrates absence of the thymic shadow in all patients with SCID (except for those with SCID due to coronin 1A deficiency).[4][40][Figure caption and citation for the preceding image starts]: Chest x-ray of an infant depicting an absent thymic shadow; infants with SCID may be athymic at time of presentationChildren's Hospital of Wisconsin, Department of Radiology; used with permission [Citation ends].
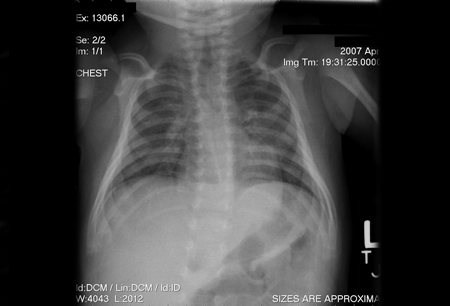 [Figure caption and citation for the preceding image starts]: Chest x-ray of an infant depicting a normal thymic shadowChildren's Hospital of Wisconsin, Department of Radiology; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Chest x-ray of an infant depicting a normal thymic shadowChildren's Hospital of Wisconsin, Department of Radiology; used with permission [Citation ends]. This observation supports the diagnosis of SCID, although a reduction in thymus size may also be caused by thymectomy, chronic inflammation, use of corticosteroids, and stress.[4]
This observation supports the diagnosis of SCID, although a reduction in thymus size may also be caused by thymectomy, chronic inflammation, use of corticosteroids, and stress.[4]
Patients with adenosine deaminase deficiency SCID may demonstrate anterior rib abnormalities on x-ray (i.e., cupping and fraying of the costochondral junctions).[41]
Ultrasound, computed tomography scan, or magnetic resonance imaging may also demonstrate the absence of a thymus.[Figure caption and citation for the preceding image starts]: Chest x-ray of an infant depicting a normal thymic shadowChildren's Hospital of Wisconsin, Department of Radiology; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Chest x-ray of an infant depicting an absent thymic shadow; infants with SCID may be athymic at time of presentationChildren's Hospital of Wisconsin, Department of Radiology; used with permission [Citation ends].
Other investigations
In infants with suspected SCID with CMV infection, regular fundoscopic evaluation for CMV retinitis should be performed.
In suspected cases of purine metabolic defects (e.g., adenosine deaminase deficiency and purine nucleoside phosphorylase deficiency), enzyme levels and associated lymphotoxic metabolites (e.g., deoxy-ATP) should be measured.[34] Purine nucleoside phosphorylase deficiency can be associated with decreased serum uric acid (<59 micromol/L [<1 mg/dL]).
Functional testing of the immune system should be performed using T-cell proliferation studies, if there are a measurable number of T cells in peripheral blood. Patients with SCID show an absent or diminished response to mitogens, including PHA and concanavalin A.[34] Consider postponing T-cell proliferation studies if T cells are absent.
Polymerase chain reaction-based viraemia testing for HIV-1, CMV, and EBV should be performed in all patients with suspected SCID. Serological studies for viral infections should not be used, as transplacentally acquired maternal antibodies may lead to false-positive results.
In patients with suspected defects in genes involved in DNA repair and recombination (e.g., Artemis/DCLRE1C, DNA ligase IV, and Cernunnos/XLF), radiation sensitivity testing of fibroblast cultures might be performed.
Genetic testing
DNA testing to identify the underlying genetic aetiology of SCID should be performed, but should not delay treatment when the diagnosis of SCID is clear. Patients with SCID due to defects in genes involved in DNA repair and recombination should not receive standard conditioning regimens for their haematopoietic stem cell transplantation, and so genetic testing or radiation sensitivity testing should be performed if feasible.[31]
Use of this content is subject to our disclaimer