Muscle and cutaneous involvement develop simultaneously in approximately 60% of patients, although in 30% skin lesions precede muscle weakness.[64]Sontheimer R, Provost T. Cutaneous manifestations of rheumatic diseases. 2nd ed. Philidelphia: Lippencott, William & Wilkins; 2004. In some patients the delay between cutaneous lesions and symptoms of muscle weakness may be several months.[65]Rockerbie NR, Woo TY, Callen JP, et al. Cutaneous changes of dermatomyositis precede muscle weakness. J Am Acad Dermatol. 1989 Apr;20(4):629-32.
http://www.ncbi.nlm.nih.gov/pubmed/2715409?tool=bestpractice.com
Between 10% and 20% of patients have typical skin lesions but never develop clinically apparent muscle disease.[2]Gerami P, Schope JM, McDonald L, et al. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis sine myositis): a missing link within the spectrum of idiopathic inflammatory myopathies. J Am Acad Dermatol. 2006 Apr;54(4):597-613.
http://www.ncbi.nlm.nih.gov/pubmed/16546580?tool=bestpractice.com
History
The cardinal musculoskeletal symptom is subacute proximal muscle weakness affecting the arms and legs. Patients report difficulty rising from a chair, climbing stairs, and washing or combing their hair. Myalgia is present in <30% of patients.[3]Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003 Sep 20;362(9388):971-82.
http://www.ncbi.nlm.nih.gov/pubmed/14511932?tool=bestpractice.com
Distal motor weakness is a rare and late feature and raises the possibility of inclusion body myositis as an alternative diagnosis. Extraocular muscles are not affected.[54]Dalakas MC. Polymyositis, dermatomyositis and inclusion-body myositis. N Engl J Med. 1991 Nov 21;325(21):1487-98.
http://www.ncbi.nlm.nih.gov/pubmed/1658649?tool=bestpractice.com
Patients may present with a variety of cutaneous manifestations (see Physical examination).
Patients may describe severe photosensitivity with blistering and pruritus.[66]Cheong WK, Hughes GR, Norris PG, et al. Cutaneous photosensitivity in dermatomyositis. Br J Dermatol. 1994 Aug;131(2):205-8.
http://www.ncbi.nlm.nih.gov/pubmed/7917983?tool=bestpractice.com
[67]Shirani Z, Kucenic MJ, Carroll CL, et al. Pruritus in adult dermatomyositis. Clin Exp Dermatol. 2004 May;29(3):273-6.
http://www.ncbi.nlm.nih.gov/pubmed/15115510?tool=bestpractice.com
Other less common symptoms reported by the patient may include:[3]Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003 Sep 20;362(9388):971-82.
http://www.ncbi.nlm.nih.gov/pubmed/14511932?tool=bestpractice.com
In malignancy-associated DM, patients may also report a range of symptoms relating to the underlying malignancy.
Physical examination
The clinical feature that differentiates DM from other inflammatory myopathies is the presence of characteristic skin involvement.[1]Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. 1975 Feb 20;292(8):403-7.
http://www.ncbi.nlm.nih.gov/pubmed/1089199?tool=bestpractice.com
Cutaneous manifestations
Heliotrope rash: periorbital dusky violaceous erythema of the eyelids often associated with periorbital oedema.
Gottron's papules: pathognomonic of DM. Violaceous flat-topped papules and plaques on the dorsal surfaces of the knuckles, and more rarely the wrists, elbows, knees, and malleoli.[Figure caption and citation for the preceding image starts]: Macular erythematous patches (Gottron’s papules) over the dorsal surface of the hands, especially over the metacarpophalangeal and proximal interphalangeal jointsAdapted from BMJ Case Reports 2009 [doi:10.1136/bcr.06.2009.2027] Copyright © 2009 by the BMJ Publishing Group Ltd [Citation ends].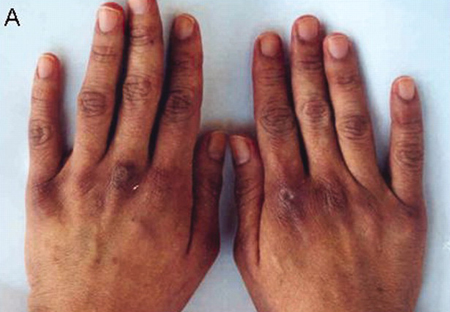 [Figure caption and citation for the preceding image starts]: Macular erythematous patches (Gottron’s papules) over the elbowsAdapted from BMJ Case Reports 2009 [doi:10.1136/bcr.06.2009.2027] Copyright © 2009 by the BMJ Publishing Group Ltd [Citation ends].
[Figure caption and citation for the preceding image starts]: Macular erythematous patches (Gottron’s papules) over the elbowsAdapted from BMJ Case Reports 2009 [doi:10.1136/bcr.06.2009.2027] Copyright © 2009 by the BMJ Publishing Group Ltd [Citation ends]. [Figure caption and citation for the preceding image starts]: Macular erythematous patches (Gottron’s papules) over the kneesAdapted from BMJ Case Reports 2009 [doi:10.1136/bcr.06.2009.2027] Copyright © 2009 by the BMJ Publishing Group Ltd [Citation ends].
[Figure caption and citation for the preceding image starts]: Macular erythematous patches (Gottron’s papules) over the kneesAdapted from BMJ Case Reports 2009 [doi:10.1136/bcr.06.2009.2027] Copyright © 2009 by the BMJ Publishing Group Ltd [Citation ends].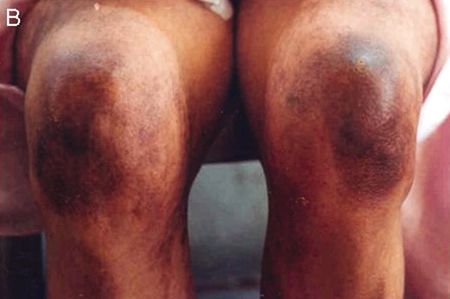
Macular violaceous erythemas: classic distributions include V sign (neck and upper chest area), shawl sign (nape of neck, posterior shoulders, and upper back), Gottron's sign (knuckles, elbows, and knees). Atrophic areas with varying hypo-pigmentation and hyper-pigmentation, telangiectasis, and scaling (poikiloderma vasculare atrophicans) may occur in areas of chronic macular violaceous erythema.
Hyper-keratosis, scaling, and fissuring of palms of hands and palmar aspect of fingers: commonly referred to as 'mechanic's' hands and associated with anti-synthetase syndrome.[45]Yoshifuji H, Fujii T, Kobayashi S, et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity. 2006 May;39(3):233-41.
http://www.ncbi.nlm.nih.gov/pubmed/16769657?tool=bestpractice.com
Cutaneous calcinosis: seen in 30% to 70% of juvenile DM; rare in adult DM and associated with anti-NXP2 antibodies.[48]Tansley SL, Betteridge ZE, Shaddick G, et al. Calcinosis in juvenile dermatomyositis is influenced by both anti-NXP2 autoantibody status and age at disease onset. Rheumatology (Oxford). 2014 Dec;53(12):2204-8.
https://academic.oup.com/rheumatology/article/53/12/2204/1804183
http://www.ncbi.nlm.nih.gov/pubmed/24987158?tool=bestpractice.com
[68]Blane CE, White SJ, Braunstein EM, et al. Patterns of calcification in childhood dermatomyositis. AJR Am J Roentgenol. 1984 Feb;142(2):397-400.
http://www.ajronline.org/doi/pdf/10.2214/ajr.142.2.397
http://www.ncbi.nlm.nih.gov/pubmed/6607616?tool=bestpractice.com
[69]Cohen MG, Nash P, Webb J. Calcification is rare in adult-onset dermatopolymyositis. Clin Rheumatol. 1986 Dec;5(4):512-6.
http://www.ncbi.nlm.nih.gov/pubmed/3816100?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Macular erythematous patches (Gottron’s papules) over the toes with dystrophic cuticlesAdapted from BMJ Case Reports 2009 [doi:10.1136/bcr.06.2009.2027] Copyright © 2009 by the BMJ Publishing Group Ltd [Citation ends].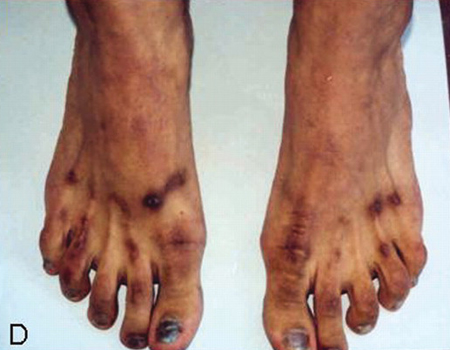
Examination of the nails may identify periungual erythema, nail-fold capillary dilation with occasional vascular drop out, and cuticular overgrowth (ragged cuticles). Although these nail changes can be seen in other connective tissue diseases, they occur most commonly in DM.[70]Ohtsuka T. The relation between nailfold bleeding and capillary microscopic abnormality in patients with connective tissue diseases. Int J Dermatol. 998 Jan;37(1):23-6.
http://www.ncbi.nlm.nih.gov/pubmed/9522233?tool=bestpractice.com
[71]Maricq HR, LeRoy EC. Patterns of finger capillary abnormalities in connective tissue disease by "wide-field" microscopy. Arthritis Rheum. 1973 Sep-Oct;16(5):619-28.
http://www.ncbi.nlm.nih.gov/pubmed/4742842?tool=bestpractice.com
Skin involvement often causes significant morbidity and can persist despite good control of muscle disease.[72]Quain RD, Werth VP. Management of cutaneous dermatomyositis: current therapeutic options. Am J Clin Dermatol. 2006;7(6):341-51.
http://www.ncbi.nlm.nih.gov/pubmed/17173468?tool=bestpractice.com
Muscle involvement
Symmetrical proximal muscle weakness is present in patients with muscle involvement; the severity varies between patients. Muscle tenderness is present in approximately 50% of patients.
Weakness of the neck flexors can result in difficulty raising the head, and bulbar involvement may lead to hoarseness, dysphonia, or nasal regurgitation.
Breathlessness may be observed during examination. When due to respiratory muscle weakness, chest expansion and air entry may be reduced.
Muscle atrophy can be seen in long-standing disease, but severe muscle atrophy is not common and alternative diagnoses should be considered when it is present.[73]Klippel JH, Dieppe PA. Rheumatology. 2nd ed. London, UK: Mosby; 1997.
Systemic manifestations
Inflammatory polyarthritis may occur and, when present, has a similar distribution to rheumatoid arthritis.[74]Schumacher HR, Schimmer B, Gordon GV, et al. Articular manifestations of polymyositis and dermatomyositis. Am J Med. 1979 Aug;67(2):287-92.
http://www.ncbi.nlm.nih.gov/pubmed/463935?tool=bestpractice.com
Joint involvement is more common when DM is part of an overlap with other connective tissue diseases. Severe deforming arthritis is more likely to be a feature of polymyositis.[75]Bunch TW, O'Duffy JD, McLeod RA. Deforming arthritis of the hands in polymyositis. Arthritis Rheum. 1976 Mar-Apr;19(2):243-8.
http://www.ncbi.nlm.nih.gov/pubmed/1259796?tool=bestpractice.com
In the presence of associated interstitial lung disease, fine bilateral crepitations may be heard on auscultation.[76]Marie I, Hatron PY, Hachulla E, et al. Pulmonary involvement in polymyositis and dermatomyositis. J Rheumatol. 1998 Jul;25(7):1336-43.
http://www.ncbi.nlm.nih.gov/pubmed/9676766?tool=bestpractice.com
Signs of cardiac failure can be present (tachycardia, raised jugular venous pressure, gallop rhythm, and oedema) when there is cardiac involvement; however, this is uncommon, and is a poor prognostic indicator.[77]Gonzalez-Lopez L, Gamez-Nava JI, Sanchez L, et al. Cardiac manifestations in dermato-polymyositis. Clin Exp Rheumatol. 1996 Jul-Aug;14(4):373-9.
http://www.ncbi.nlm.nih.gov/pubmed/8871835?tool=bestpractice.com
[78]Riemekasten G, Opitz C, Audring H, et al. Beware of the heart: the multiple picture of cardiac involvement in myositis. Rheumatology (Oxford). 1999 Nov;38(11):1153-7.
http://rheumatology.oxfordjournals.org/cgi/content/full/38/11/1153
http://www.ncbi.nlm.nih.gov/pubmed/10556273?tool=bestpractice.com
[79]Haupt HM, Hutchins GM. The heart and cardiac conduction system in polymyositis-dermatomyositis: a clinicopathologic study of 16 autopsied patients. Am J Cardiol. 1982 Nov;50(5):998-1006.
http://www.ncbi.nlm.nih.gov/pubmed/7137049?tool=bestpractice.com
[80]Stern R, Godbold JH, Chess Q, et al. ECG abnormalities in polymyositis. Arch Intern Med. 1984 Nov;144(11):2185-9.
http://www.ncbi.nlm.nih.gov/pubmed/6497519?tool=bestpractice.com
Raynaud's phenomenon may be observed but is more common in overlap DM.[81]Leteurtre E, Hachulla E, Janin A, et al. Vascular manifestations of dermatomyositis and polymyositis. Clinical, capillaroscopic and histological aspects [in French]. Rev Med Interne. 1994;15(12):800-7.
http://www.ncbi.nlm.nih.gov/pubmed/7863114?tool=bestpractice.com
Diagnostic investigations
The initial order of investigations will be influenced by whether the patient presents with cutaneous symptoms, muscle symptoms, or both.
Patients with clinically amyopathic dermatomyositis (CADM) need evaluation for subclinical muscle disease. This should be sought by performing creatine kinase (CK) measurement, electromyogram, muscle biopsy, and magnetic resonance imaging (MRI).[82]Jorizzo J. Dermatomyositis: practical aspects. Arch Dermatol. 2002 Jan;138(1):114-6.
http://www.ncbi.nlm.nih.gov/pubmed/11790176?tool=bestpractice.com
Serum creatine kinase (CK) and serum aldolase
As the most sensitive muscle enzyme test, CK should be ordered for any patient with suspected DM, even if the patient presents with only cutaneous involvement, as CK may be elevated in CADM.[2]Gerami P, Schope JM, McDonald L, et al. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis sine myositis): a missing link within the spectrum of idiopathic inflammatory myopathies. J Am Acad Dermatol. 2006 Apr;54(4):597-613.
http://www.ncbi.nlm.nih.gov/pubmed/16546580?tool=bestpractice.com
[83]Vignos PJ, Goldwyn J. Evaluation of laboratory tests in diagnosis and management of polymyositis. Am J Med Sci. 1972 Apr;263(4):291-308.
http://www.ncbi.nlm.nih.gov/pubmed/5031996?tool=bestpractice.com
Serum aldolase is a glycolytic pathway enzyme found in all tissues but mainly skeletal muscle, brain, and liver. It is not as specific or sensitive as CK for muscle disease, but is occasionally elevated in myositis when CK is normal.
Muscle biopsy
Muscle biopsy should be performed in all cases of suspected DM. In symptomatic patients, the biopsy should be taken from a weak, but not severely atrophied, muscle. Quadriceps and deltoid are common sites.
Open surgical biopsy provides a larger specimen compared with closed needle biopsy. Biopsy using a conchotome also yields useful specimens.[84]Dorph C, Nennesmo I, Lundberg IE. Percutaneous conchotome muscle biopsy: a useful diagnostic and assessment tool. J Rheumatol. 2001 Jul;28(7):1591-9.
http://www.ncbi.nlm.nih.gov/pubmed/11469467?tool=bestpractice.com
Correct processing and interpretation by a laboratory experienced in muscle histology is essential for correct diagnosis.
Electromyography (EMG)
EMG is recommended, but it is not essential, for diagnosis if raised CK and typical muscle biopsy findings are present. The EMG abnormalities are not specific but are seen more frequently in idiopathic inflammatory myopathies.[85]Barkhaus PE, Nandedkar SD, Sanders DB. Quantitative EMG in inflammatory myopathy. Muscle Nerve. 1990 Mar;13(3):247-53.
http://www.ncbi.nlm.nih.gov/pubmed/2320046?tool=bestpractice.com
[86]Uncini A, Lange DJ, Lovelace RE, et al. Long-duration polyphasic motor unit potentials in myopathies: a quantitative study with pathological correlation. Muscle Nerve. 1990 Mar;13(3):263-7.
http://www.ncbi.nlm.nih.gov/pubmed/2320048?tool=bestpractice.com
Similar patterns may be seen in toxic, infectious, or metabolic myopathies.
Skin biopsy
Skin biopsy is required to confirm diagnosis in CADM, where overlap connective tissue disease may be present, or in cases where the cutaneous diagnosis is in doubt. In patients who present with classic DM and the diagnosis is confirmed by raised muscle enzymes and muscle biopsy, skin biopsy may not be required.
Antinuclear antibody (ANA)
Positive in approximately 80% of patients.[87]Reichlin M, Arnett FC. Multiplicity of antibodies in myositis sera. Arthritis Rheum. 1984 Oct;27(10):1150-6.
http://www.ncbi.nlm.nih.gov/pubmed/6386003?tool=bestpractice.com
Non-specific and commonly positive in other connective tissue diseases where myositis may be a feature. Further testing for autoantibodies against specific nuclear antigens (e.g., anti-double-stranded DNA, anti-Ro, anti-La, anti-Sm, anti-ribonucleoprotein [RNP]) is useful to help differentiate between DM and other connective tissue diseases, especially cutaneous lupus, which has a similar biopsy appearance.
Myositis-specific antibodies (MSAs) and myositis-associated antibodies (MAAs)
MSAs and MAAs are found in most patients with DM and are associated with clinical subsets that have varying presentations, clinical courses, and therapeutic responses.[24]DeWane ME, Waldman R, Lu J. Dermatomyositis: clinical features and pathogenesis. J Am Acad Dermatol. 2020 Feb;82(2):267-81.
http://www.ncbi.nlm.nih.gov/pubmed/31279808?tool=bestpractice.com
[88]Merlo G, Clapasson A, Cozzani E, et al. Specific autoantibodies in dermatomyositis: a helpful tool to classify different clinical subsets. Arch Dermatol Res. 2017 Mar;309(2):87-95.
http://www.ncbi.nlm.nih.gov/pubmed/27928683?tool=bestpractice.com
[89]Alenzi FM. Myositis specific autoantibodies: a clinical perspective. Open Access Rheumatol. 2020 Jan 14;12:9-14.
https://www.dovepress.com/myositis-specific-autoantibodies-a-clinical-perspective-peer-reviewed-fulltext-article-OARRR
http://www.ncbi.nlm.nih.gov/pubmed/32021502?tool=bestpractice.com
They are increasingly used in clinical practice, although are not widely available.[50]Kohsaka H, Mimori T, Kanda T, et al. Treatment consensus for management of polymyositis and dermatomyositis among rheumatologists, neurologists and dermatologists. Mod Rheumatol. 2019 Jan;29(1):1-19.
http://www.ncbi.nlm.nih.gov/pubmed/30565491?tool=bestpractice.com
Most are directed against cytoplasmic RNA-protein complexes.[46]Hengstman GJ, van Engelen BG, van Venrooij WJ. Myositis specific autoantibodies: changing insights in pathophysiology and clinical associations. Curr Opin Rheumatol. 2004 Nov;16(6):692-9.
http://www.ncbi.nlm.nih.gov/pubmed/15577606?tool=bestpractice.com
Anti-Mi-2 antibodies are directed against a helicase involved in transcriptional activation.[90]Seelig HP, Moosbrugger I, Ehrfeld H, et al. The major dermatomyositis-specific autoantigen is a presumed helicase involved in transcriptional activation. Arthritis Rheum. 1995 Oct;38(10):1389-99.
http://www.ncbi.nlm.nih.gov/pubmed/7575689?tool=bestpractice.com
They are strongly associated with the classical presentation of DM but are found in only approximately 10% of patients.[91]Targoff IN, Reichlin M. The association between Mi-2 antibodies and dermatomyositis. Arthritis Rheum. 1985 Jul;28(7):796-803.
http://www.ncbi.nlm.nih.gov/pubmed/2409985?tool=bestpractice.com
This subset of patients have the highest response to steroid treatment and the lowest incidence of malignancy. Anti‐U1 RNP and anti‐Ku are found in overlap syndromes that present similarly to anti-Mi-2 positive DM.[50]Kohsaka H, Mimori T, Kanda T, et al. Treatment consensus for management of polymyositis and dermatomyositis among rheumatologists, neurologists and dermatologists. Mod Rheumatol. 2019 Jan;29(1):1-19.
http://www.ncbi.nlm.nih.gov/pubmed/30565491?tool=bestpractice.com
Anti‐transcription intermediary factor 1 gamma (TIF1-gamma) is the most commonly isolated antibody.[88]Merlo G, Clapasson A, Cozzani E, et al. Specific autoantibodies in dermatomyositis: a helpful tool to classify different clinical subsets. Arch Dermatol Res. 2017 Mar;309(2):87-95.
http://www.ncbi.nlm.nih.gov/pubmed/27928683?tool=bestpractice.com
It is strongly associated with malignancy and therefore an intensive search for cancer and careful follow‐up is recommended.[45]Yoshifuji H, Fujii T, Kobayashi S, et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity. 2006 May;39(3):233-41.
http://www.ncbi.nlm.nih.gov/pubmed/16769657?tool=bestpractice.com
[92]Best M, Molinari N, Chasset F, et al. Use of anti-transcriptional intermediary factor-1 gamma autoantibody in identifying adult dermatomyositis patients with cancer: a systematic review and meta-analysis. Acta Derm Venereol. 2019 Mar 1;99(3):256-62.
https://www.medicaljournals.se/acta/content/html/10.2340/00015555-3091
http://www.ncbi.nlm.nih.gov/pubmed/30460368?tool=bestpractice.com
The rash is often refractory to treatment.
Anti-signal recognition particle (SRP) antibodies are found almost exclusively in polymyositis and occasionally in DM. When present they are associated with acute severe, treatment-resistant necrotising myositis.[46]Hengstman GJ, van Engelen BG, van Venrooij WJ. Myositis specific autoantibodies: changing insights in pathophysiology and clinical associations. Curr Opin Rheumatol. 2004 Nov;16(6):692-9.
http://www.ncbi.nlm.nih.gov/pubmed/15577606?tool=bestpractice.com
Immunosuppressive drugs, intravenous immunoglobulin, or rituximab are often required at an early disease stage.[93]Hengstman GJ, ter Laak HJ, Vree Egberts WT, et al. Anti-signal recognition particle autoantibodies: marker of a necrotising myopathy. Ann Rheum Dis. 2006 Dec;65(12):1635-8.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1798474
http://www.ncbi.nlm.nih.gov/pubmed/16679430?tool=bestpractice.com
Anti-Jo-1 and other anti-aminoacyl-tRNA synthetase antibodies (anti-ARS antibodies; including anti‐PL‐7, anti‐PL‐12, anti‐EJ, anti‐OJ, and anti‐KS) result in a distinct clinical picture known as anti-synthetase syndrome. The main clinical features include interstitial lung disease, fever, myositis, polyarthritis, mechanic's hands, and Raynaud's phenomenon. Anti-Jo-1 is associated with a higher frequency of myositis, whereas the other anti-ARS antibodies are associated with higher frequency of interstitial lung disease. Anti-synthetase syndrome is often steroid resistant and concomitant use of immunosuppressive drugs required.[45]Yoshifuji H, Fujii T, Kobayashi S, et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity. 2006 May;39(3):233-41.
http://www.ncbi.nlm.nih.gov/pubmed/16769657?tool=bestpractice.com
Anti-melanoma differentiation-associated gene 5 (MDA5; also known as CADM‐140) is specific to CADM and is strongly associated with a rapidly progressive interstitial lung disease with poor prognosis. Tender palm papules and diffuse alopecia may be present. Patients often require treatment with immunosuppressive drugs as well as high‐dose systemic steroids from an early disease stage.[47]Sato S, Hirakata M, Kuwana M, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum. 2005 May;52(5):1571-6.
https://onlinelibrary.wiley.com/doi/10.1002/art.21023
http://www.ncbi.nlm.nih.gov/pubmed/15880816?tool=bestpractice.com
Anti-NXP2 is strongly associated with juvenile DM with a high proportion suffering from cutaneous calcinosis due to dystrophic calcification.[48]Tansley SL, Betteridge ZE, Shaddick G, et al. Calcinosis in juvenile dermatomyositis is influenced by both anti-NXP2 autoantibody status and age at disease onset. Rheumatology (Oxford). 2014 Dec;53(12):2204-8.
https://academic.oup.com/rheumatology/article/53/12/2204/1804183
http://www.ncbi.nlm.nih.gov/pubmed/24987158?tool=bestpractice.com
Anti-PM-Scl antibodies have been found in patients with overlapping myositis and scleroderma.[49]Jablonska S, Blaszyk M. Scleromyositis (scleroderma/polimyositis overlap) is an entity. J Eur Acad Dermatol Venereol. 2004 May;18(3):265-6.
http://www.ncbi.nlm.nih.gov/pubmed/15096133?tool=bestpractice.com
Muscle magnetic resonance imaging (MRI)
MRI may improve biopsy yield by identifying affected muscles to target for biopsy.[94]Tomasova Studynkova J, Charvat F, Jarosova K, et al. The role of MRI in the assessment of polymyositis and dermatomyositis. Rheumatology (Oxford). 2007 Jul;46(7):1174-9.
http://rheumatology.oxfordjournals.org/cgi/content/full/46/7/1174
http://www.ncbi.nlm.nih.gov/pubmed/17500079?tool=bestpractice.com
MRI findings are not specific and biopsy is required for diagnosis.[95]Adams EM, Chow CK, Premkumar A, et al. The idiopathic inflammatory myopathies: spectrum of MR imaging findings. Radiographics. 1995 May;15(3):563-74.
http://radiographics.rsna.org/content/15/3/563.full.pdf
http://www.ncbi.nlm.nih.gov/pubmed/7624563?tool=bestpractice.com
Serial MRI measurements may also be performed to assess response to treatment.
Screening for major organ involvement and determining prognosis
Patients with classic dermatomyositis (CDM) or amyopathic dermatomyositis (ADM) should be investigated for occult malignancy and other disease manifestations such as myocardial involvement and interstitial lung disease.[2]Gerami P, Schope JM, McDonald L, et al. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis sine myositis): a missing link within the spectrum of idiopathic inflammatory myopathies. J Am Acad Dermatol. 2006 Apr;54(4):597-613.
http://www.ncbi.nlm.nih.gov/pubmed/16546580?tool=bestpractice.com
[82]Jorizzo J. Dermatomyositis: practical aspects. Arch Dermatol. 2002 Jan;138(1):114-6.
http://www.ncbi.nlm.nih.gov/pubmed/11790176?tool=bestpractice.com
[96]Dawkins MA, Jorizzo JL, Walker FO, et al. Dermatomyositis: a dermatology-based case series. J Am Acad Dermatol. 1998 Mar;38(3):397-404.
http://www.ncbi.nlm.nih.gov/pubmed/9520020?tool=bestpractice.com
[97]Stonecipher MR, Jorizzo JL, White WL, et al. Cutaneous changes of dermatomyositis in patients with normal muscle enzymes: dermatomyositis sine myositis? J Am Acad Dermatol. 1993 Jun;28(6):951-6.
http://www.ncbi.nlm.nih.gov/pubmed/8496459?tool=bestpractice.com
Electrocardiogram (ECG)
Should be performed in all patients. Around 30% of patients with idiopathic inflammatory myopathies may have ECG abnormalities.[80]Stern R, Godbold JH, Chess Q, et al. ECG abnormalities in polymyositis. Arch Intern Med. 1984 Nov;144(11):2185-9.
http://www.ncbi.nlm.nih.gov/pubmed/6497519?tool=bestpractice.com
Patients with normal resting ECG but symptoms of palpitations or syncope merit further investigation with ambulatory ECG.
Echocardiography
Should be performed in all patients. Myocardial involvement severe enough to cause congestive cardiac failure is rare (<5% of patients), but when present indicates poor prognosis.[77]Gonzalez-Lopez L, Gamez-Nava JI, Sanchez L, et al. Cardiac manifestations in dermato-polymyositis. Clin Exp Rheumatol. 1996 Jul-Aug;14(4):373-9.
http://www.ncbi.nlm.nih.gov/pubmed/8871835?tool=bestpractice.com
Chest x-ray
Required in all patients to evaluate respiratory involvement and to screen for malignancy. In early interstitial lung disease, radiographical changes may be minimal or non-specific and interstitial lung disease may be missed if further respiratory investigations are not performed.
Cardiac troponin I
A sensitive and specific marker of cardiac damage, cardiac troponin I is used to screen asymptomatic patients at presentation with idiopathic inflammatory myopathies.[98]Hughes M, Lilleker JB, Herrick AL, et al. Cardiac troponin testing in idiopathic inflammatory myopathies and systemic sclerosis-spectrum disorders: biomarkers to distinguish between primary cardiac involvement and low-grade skeletal muscle disease activity. Ann Rheum Dis. 2015 May;74(5):795-8.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4589894
http://www.ncbi.nlm.nih.gov/pubmed/25732174?tool=bestpractice.com
Pulmonary function tests (PFTs)
Required in all patients to evaluate for respiratory muscle weakness and interstitial lung disease.[99]American College of Rheumatology. 2023 American College of Rheumatology (ACR) guideline for the screening and monitoring of interstitial lung disease in people with systemic autoimmune rheumatic disease. Aug 2023 [internet publication].
https://assets.contentstack.io/v3/assets/bltee37abb6b278ab2c/blt7e2cadfc7bc986fb/interstitial-lung-disease-guideline-summary-screening-monitoring-2023.pdf
High-resolution CT of chest
Should be performed in all patients to evaluate for interstitial lung disease.[99]American College of Rheumatology. 2023 American College of Rheumatology (ACR) guideline for the screening and monitoring of interstitial lung disease in people with systemic autoimmune rheumatic disease. Aug 2023 [internet publication].
https://assets.contentstack.io/v3/assets/bltee37abb6b278ab2c/blt7e2cadfc7bc986fb/interstitial-lung-disease-guideline-summary-screening-monitoring-2023.pdf
More sensitive than chest x-ray in detecting interstitial lung disease and provides information on prognosis not provided by chest x-ray. The presence of ground glass opacification is associated with a better prognosis compared with honeycomb fibrotic changes.[100]Padley SP, Hansell DM, Flower CD, et al. Comparative accuracy of high resolution computed tomography and chest radiography in the diagnosis of chronic diffuse infiltrative lung disease. Clin Radiol. 1991 Oct;44(4):222-6.
http://www.ncbi.nlm.nih.gov/pubmed/1959296?tool=bestpractice.com
[101]Bonnefoy O, Ferretti G, Calaque O, et al. Serial chest CT findings in interstitial lung disease associated with polymyositis-dermatomyositis. Eur J Radiol. 2004 Mar;49(3):235-44.
http://www.ncbi.nlm.nih.gov/pubmed/14962653?tool=bestpractice.com
Barium swallow or video-fluoroscopic assessment of swallow
Should be performed in patients with dysphagia, nasal regurgitation of fluid, or dysphonia.
Silent aspiration may also occur and further assessment is indicated in patients presenting with lower respiratory tract infection.[102]de Merieux P, Verity MA, Clements PJ, et al. Esophageal abnormalities and dysphagia in polymyositis and dermatomyositis. Arthritis Rheum. 1983 Aug;26(8):961-8.
http://www.ncbi.nlm.nih.gov/pubmed/6882490?tool=bestpractice.com
Investigations for associated malignancy
Full blood count, serum ferritin, serum biochemistry screen
Urinalysis
Faecal occult blood testing
Chest x-ray
Women: mammography, CA-125, and pelvic ultrasound
Men: prostate-specific antigen
CT chest/abdomen/pelvis
Other investigations (e.g., gastrointestinal endoscopy) guided by symptoms and abnormal results.[103]Amoura Z, Duhaut P, Huong DL, et al. Tumor antigen markers for the detection of solid cancers in inflammatory myopathies. Cancer Epidemiol Biomarkers Prev. 2005 May;14(5):1279-82.
http://cebp.aacrjournals.org/content/14/5/1279.full
http://www.ncbi.nlm.nih.gov/pubmed/15894686?tool=bestpractice.com
[104]Provost TT, Flynn JA. Dermatomyositis. In: Provost TT, Flynn JA, eds. Cutaneous medicine. Hamilton, Ontario, Canada: B.C. Decker Inc; 2001:82-103.[105]Drake LA, Dinehart SM, Farmer ER, et al; American Academy of Dermatology. Guidelines of care for dermatomyositis. J Am Acad Dermatol. 1996 May;34(5 Pt 1):824-9.
http://www.ncbi.nlm.nih.gov/pubmed/8632081?tool=bestpractice.com
[106]Sparsa A, Liozon E, Herrmann F, et al. Routine vs extensive malignancy search for adult dermatomyositis and polymyositis: a study of 40 patients. Arch Dermatol. 2002 Jul;138(7):885-90.
http://archderm.ama-assn.org/cgi/content/full/138/7/885
http://www.ncbi.nlm.nih.gov/pubmed/12071815?tool=bestpractice.com