Approach
The key step is a carefully elicited history.
Identification of risk factors
Risk factors strongly associated with Huntington's disease include the presence of a positive family history and the presence of an expanded CAG repeat length at the N-terminal of the huntingtin gene. Occasionally, no family history for Huntington's disease is found. In these cases, consideration should be given to false paternity, early death of an affected parent, or CAG repeat expansion from an unaffected parent.
Cognitive impairment
The earliest symptoms in Huntington's disease relate to loss of executive functions, and these typically are best captured in discussions about the patient's work or home life. Common findings include:
Concentration impairment (performance at work or school may be impaired)
Task apathy and anxiety
Increased errors in complex functions
Assumption of responsibilities by colleagues or family members
Slips, misjudgements, or motor vehicle accidents.
Although not considered a typical aspect of the physical examination, comparing patients with spouses or siblings well matched for education or IQ is often informative.
Behavioural features
Behavioural manifestations commonly include:[23]
Irritability
Impulsivity
Lack of attention to appearance or hygiene
Personality changes. Although sometimes difficult to categorise, these are often noticed, sometimes as changes in habits and interests. If there is denial, it may help to interview family members separately.[7][24]
Patients often seem a little disinhibited or unusually anxious. Formal bedside mental status tests, such as the mini-mental state examination, are often normal, particularly if the patient is well educated. More helpful is recognition of occasional inappropriateness or lack of focus of the patient, particularly compared with the spouse or accompanying family member.
Motor features
Motor features may include:
Chorea
Twitching/restlessness: family members may note the presence of these features and changes in coordination
Bradykinesia and rigidity.
The most useful findings on examination relate to chorea.[7][24][25] Patients are sometimes able to suppress involuntary movements, so it is helpful to spend a few minutes specifically looking for them. Having the patient seated, shoes off, with eyes closed, reciting serial 7s with hands outstretched is useful. Random movements of fingers and toes, with occasional peculiar postures of hands, torso, or limbs, and odd facial expressions are typical of early Huntington's disease, as are impaired rhythm and rate of tapping of the index finger on the thumb, and motor impersistence (the inability to keep the tongue fully protruded).
The demonstration of chorea in verbal terms is difficult, but an electromyogram (EMG) recording of a finger muscle in an affected patient shows the random nature of this type of movement disorder. Slowed rapid (saccadic) eye movements may be detected. This feature is often prominent in juvenile Huntington's disease. Difficulty with tandem walking may be seen as well.
For physicians who have had limited experience with chorea and Huntington's disease, and this includes many neurologists, referral to a movement disorders consultant or medical centre involved in Huntington's research is advisable. This is a relatively rare disorder and clinical consultation is no more time consuming than an MRI scan, yet is less costly and more informative.[Figure caption and citation for the preceding image starts]: EMG of chorea in stage I disease; recording made with standard belly tendon, surface disc electrodes over first dorsal interosseous muscleWalker FO. Huntington's disease. Lancet 2007 Jan 20;369(9557):218-28. Used with permission [Citation ends].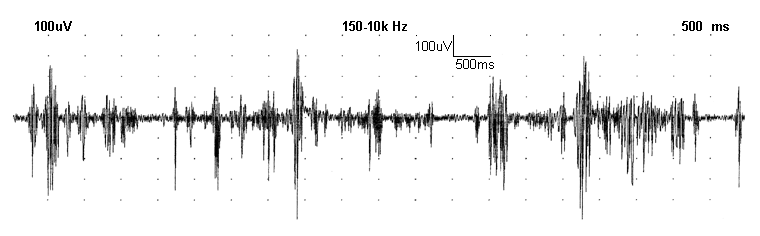
Other features
Depression may be a comorbid feature of the disease but is non-specific and is not a common initial presenting manifestation.[23][26][27][28] Obsessional behaviour and compulsions also occur, but, again, these do not usually present until later on in the course of the illness, often after the diagnosis has been made.[23]
Imaging
Imaging is usually unhelpful in the assessment of early Huntington's disease. However, atrophy of the caudate nucleus, apparent on either CT scan or MRI, is a consistent finding in later stages of the disease.[19]
Consent to further testing
Diagnosis is helpful for documenting disability, but otherwise may be unwelcome news because of the lack of disease-modifying therapy. It may be helpful to inform patients of the potential insurance and familial implications of a diagnosis, tell them that they are presenting with features that may or may not be evidence of Huntington's disease, and ask them whether they would like the diagnosis confirmed with genetic testing. Such testing can be deferred until a time of the patient's choosing and may help them decide how they wish to manage ongoing life, financial, or work issues.[7][24]
Genetic testing
If the patient has features typical of Huntington's disease, the most useful confirmatory test is CAG repeat testing. This typically takes several weeks. A positive result has many implications for the patient and family, so it is usually best to give information about Huntington's disease beforehand so that they are prepared. In presenting the results of a positive test, it is typical to arrange for the patient to come in with a spouse or supportive friend or family member. The delivery of a diagnosis can be a significant emotional event for the patient and represents a critical time for educating the family further about Huntington's disease and its genetic implications for family members and family planning. If confirmatory genetic testing is negative, it is likely the patient will need referral to a movement disorders expert to detect other possible causes of their symptoms.
Testing may also be conducted in older at-risk individuals without symptoms of Huntington's disease, in order to define the risk for the disease in their offspring. Pre-symptomatic genetic testing for at-risk family members is recommended only at medical centres specialising in such testing using published guidelines. Predictive testing for those under the age of 18 years is not recommended; individuals requesting predictive testing should have access to genetic counselling.[29][30][31][32]
Use of this content is subject to our disclaimer