Approach
A characteristic personal and family history, along with physical examination findings, raises suspicion for the diagnosis of congenital haemophilia. This is subsequently confirmed by laboratory findings. The age of presentation and bleeding frequency are influenced by the severity of the condition. Most patients are diagnosed as children. However, some patients may go undiagnosed until adulthood, particularly those with mild or even moderate haemophilia who have had no significant haemostatic challenges in their lives. Haemophilia A and B are clinically indistinguishable.
Acquired haemophilia is a rare autoimmune condition resulting from the production of auto-antibodies which inactivate factor VIII. It is often not recognised or mistaken for other acquired bleeding disorders, resulting in delayed diagnosis and management.
History
Risk factors strongly associated with congenital haemophilia include:
A family history of haemophilia, usually positive from the maternal side (uncles, cousins, grandfather)
Male sex.
Musculoskeletal bleeding is the hallmark of haemophilia. Typical history in a patient with congenital haemophilia includes recurrent or severe bleeding symptoms, or bleeding in joints or muscles. Minor mucocutaneous bleeding (e.g., epistaxis, bleeding from gums following minor dental procedures, easy bruising) as well as severe bleeding following trauma, surgery, or dental procedures, are commonly described.
Gastrointestinal bleeding and haematuria are seen in patients with congenital haemophilia. They may occur spontaneously or following trauma. Women and girls who are carriers of congenital haemophilia, particularly those with clotting factor levels in the haemophilia range, most commonly present with menorrhagia and bleeding following surgical procedures or childbirth.[25]
In the neonatal period, the following are more typical presentations of congenital haemophilia:
Intracranial bleeding that may occur spontaneously after birth, or may be precipitated by prolonged labour or instrumental delivery
Prolonged bleeding from heel prick
Prolonged bleeding following circumcision: bleeding occurs in about 50% of newborns with haemophilia who undergo circumcision.[26]
About half the cases of acquired haemophilia are associated with autoimmune conditions, malignant disease, certain drugs, or pregnancy. Unlike patients with congenital haemophilia, these patients do not have a personal or family history of bleeding episodes.
Physical examination
Typical findings on examination in patients with congenital haemophilia vary depending on the age of the patient, the severity of the type of haemophilia, and the site of bleeding. About 3% to 5% of newborn boys with severe congenital haemophilia present with intracranial haemorrhage.[27][28][29][30] Symptoms and signs are not specific, but include hypoactivity, decreased oral intake, irritability, bulging/tense fontanelle, seizures, and pallor.
Other possible signs of neonatal presentation include:
Active bleeding from a site of injury
Pain/swelling of an extremity
Distended and painful abdomen.
Typical signs of childhood or adult presentation include:
Active bleeding from a site of injury
Joint pain/swelling
Extremity pain/swelling
Decreased range of motion of an extremity
Distended and painful abdomen
Pallor
Focal neurological deficits
Haematuria.
If intracranial haemorrhage occurs in childhood, signs and symptoms may include hypoactivity, irritability, headache, vomiting, seizures, and focal neurological deficits. Although poor growth and failure to thrive may be attributed to the presence of haemophilia, they are not common signs and an alternative diagnosis should be sought.
Musculoskeletal bleeding is the hallmark of congenital haemophilia. Common bleeding sites include knee, ankle, and elbow joints, although any joint may be involved. Usual presentation includes joint pain, swelling, and decreased range of motion. Muscular bleeding may occur at any muscle, including, but not limited to:
Quadriceps
Hamstrings
Iliopsoas
Biceps
Triceps.
Usual presentation of muscular bleeding includes pain at the site, swelling, increased warmth, erythema, and limitation in range of motion. Musculoskeletal bleeding in patients with severe haemophilia (not on prophylaxis) may occur as often as once weekly. In those with moderate haemophilia, musculoskeletal bleeding may occur once a month or less, and it may occur about once a year or less in those with mild haemophilia, and usually only with significant trauma or invasive procedure/surgery.[31][32][33]
Excessive bruising or haematoma formation may also occur. Common sites are the lower extremities, although other sites may be affected. Bleeding into the iliopsoas muscle may present as severe lower abdominal, upper thigh, and/or lower back pain. Patients have pain on extension, but not on rotation, of the hip joint and typically have a characteristic gait (flexed hip, inward rotation).
Findings on examination in patients with the acquired form include extensive cutaneous purpura and signs of internal haemorrhage. Bleeding into the joints is not a prominent feature. The reason for the different bleeding pattern in acquired haemophilia remains unknown. There is no demonstrable impairment of platelet function.[Figure caption and citation for the preceding image starts]: Acute haemarthrosis of the right knee with ecchymosisPediatric Hematology Department, University of Texas Health Science Center, Houston; used with permission [Citation ends].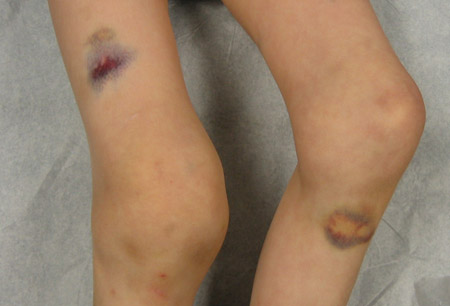 [Figure caption and citation for the preceding image starts]: Bilateral acute haemarthroses of the kneesPediatric Hematology Department, University of Texas Health Science Center, Houston; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Bilateral acute haemarthroses of the kneesPediatric Hematology Department, University of Texas Health Science Center, Houston; used with permission [Citation ends].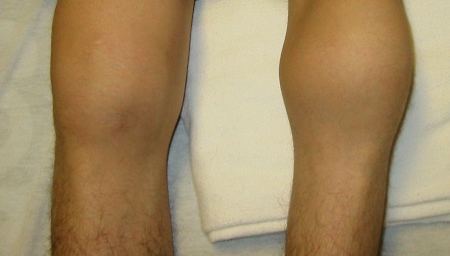 [Figure caption and citation for the preceding image starts]: Massive swelling due to acute haemarthrosis of the right kneePediatric Hematology Department, University of Texas Health Science Center, Houston; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Massive swelling due to acute haemarthrosis of the right kneePediatric Hematology Department, University of Texas Health Science Center, Houston; used with permission [Citation ends].
Initial laboratory investigations
An activated partial thromboplastin time (aPTT) should be ordered if a diagnosis of congenital haemophilia is suspected. A factor VIII and/or factor IX assay should be requested to confirm the diagnosis if the aPTT is prolonged. The aPTT may not be prolonged in mild cases (factor levels >30%), but if the diagnosis is clinically suspected, factor VIII and IX assays should be ordered. If the aPTT is prolonged, a mixing study (incubating patient plasma with normal plasma for 2 hours at 37°C [98.6°F] and aPTT repeated) may also be requested. Correction with mixing study suggests a coagulation factor deficiency, while the lack of correction suggests the presence of a coagulation inhibitor.
Diagnosis of acquired haemophilia is based on the finding of a low factor VIII level associated with the presence of a time-dependent inhibitor in the plasma. In acquired haemophilia, prolonged aPTT cannot be corrected by incubating patient plasma with normal plasma for 2 hours at 37°C (98.6°F) due to the presence of time- and temperature-dependent factor VIII inhibitor
Other tests that are indicated as part of the differential work-up include:
FBC: to rule out thrombocytopenia as a cause of bleeding and to diagnose anaemia
Prothrombin time (PT): to evaluate the extrinsic and common pathways of coagulation
Closure time/bleeding time and platelet aggregation studies: to evaluate platelet function
Von Willebrand factor studies: to rule out von Willebrand disease
Other specific factor assays: as needed, based on PT and aPTT results (for example, if the PT is prolonged, factor VII and factor V assays should be checked to rule out co-existent factor VII deficiency or the rare autosomal recessive combined factor V and VIII deficiency state; if only the aPTT is prolonged, but factors VIII and IX are normal, then factors XII and XI assays should also be checked)
Liver aminotransferases (aspartate aminotransferase [AST] and alanine aminotransferase [ALT]): to evaluate for liver dysfunction that can also contribute to prolongation of PT and aPTT
Mixing study and lupus inhibitor screen to exclude inhibitor-mediated prolonged aPTT.
Imaging and endoscopy
Imaging studies or endoscopy may be required for evaluation of acute bleeding. Investigations, as appropriate, may include:
Head CT and/or MRI: for evaluation of intracranial haemorrhage[30]
Neck CT and/or MRI: for evaluation of bleeding near the airway
Abdominal ultrasound or abdominopelvic CT scan: for evaluation of gastrointestinal bleeding or iliopsoas bleeding
Upper and/or lower endoscopy: for evaluation of gastrointestinal bleeding
Plain x-rays: as necessary, for bone evaluation. X-rays have been traditionally used to describe the clinical progression of arthropathy.[34] MRI and ultrasound may detect soft-tissue bleeding at an early stage. MRI may be helpful to evaluate whether a patient is a candidate for surgical procedures including synovectomy, joint fusion, or joint replacement.
Subsequent investigations
Can include mutation analysis, factor VIII or IX inhibitor screen, and Bethesda assay. These investigations are reserved for patients with a diagnosis of haemophilia.
Mutation analysis
Factor VIII or IX mutation analysis identifies specific genetic mutations involved in haemophilia A and B. It is performed to establish:
Diagnostic accuracy
Clinical severity
Risk of inhibitor development.
Mutation analysis is recommended for patients with a diagnosis of haemophilia A or B, affected male relatives, and female relatives at risk of being carriers.[35] It can also be used to inform antenatal diagnosis.
Mutation analysis should be carried out at specialised centres, experienced in haemophilia genetic testing. Genetic counselling should be provided before and after testing.[35]
Haemophilia B patients with genetic mutations resulting from deletions require careful observation because they may develop anaphylaxis and/or nephrotic syndrome when factor IX is infused.[36][37] The allergic reaction to factor IX generally indicates that the patient is developing or has developed a factor IX inhibitor.
Factor VIII or IX inhibitor screen
Screening detects the presence of inhibitory antibodies to infused factor VIII or IX. Indications for inhibitor screening include:[38]
Initial factor exposure
Intensive factor exposure (e.g., daily exposure for more than 5 days)
Recurrent bleeds or target joint bleeds (despite adequate clotting factor concentrate replacement therapy)
Failure to respond to adequate clotting factor concentrate replacement therapy
Lower than expected factor recovery or half-life after clotting factor concentrate replacement therapy
Suboptimal clinical or laboratory response to clotting factor concentrate replacement therapy
Before surgery
Suboptimal post-operative response to clotting factor concentrate replacement therapy.
Patients with mild haemophilia who are infrequently infused may be checked every 12 months. If patients with inhibitors have an acute bleed, treatment is complex and should be managed in a specialist centre.
About 30% of patients with haemophilia A develop inhibitors to infused factor VIII, whereas the inhibitor incidence for haemophilia B is up to 5%.[38]
Risk factors for the development of inhibitors include:[19][22][39][40][41][42][43][44][45][46][47][48]
Disease severity: more common in severe haemophilia (but may also occur in patients with mild to moderate haemophilia)
Environmental factors: treatment intensity, infection, age, type of product
Genetic mutations: in particular, those that result in absence of a gene product; immune response genes have also been implicated in inhibitor development. People with a family history of developing inhibitors to infused factor VIII and people of African-Caribbean or Hispanic descent are more likely to develop inhibitors.
No evidence has been found to support an association between the development of inhibitors and immunisation or the timing of immunisation relative to factor VIII infusion.[49][50]
Whether there is a difference in inhibitor incidence between recipients of plasma-derived and recombinant factor VIII products is under intense investigation. Some studies show no difference, while one trial of previously untransfused patients with severe haemophilia A suggested that the incidence of inhibitors was higher among those patients that had received recombinant factor VIII than in those randomised to plasma-derived factor VIII.[51][52][53][54] The US National Hemophilia Foundation advises that the development of inhibitors is a significant risk in the management of patients with previously untreated haemophilia A, and that inhibitors to factor VIII can occur with both plasma-derived and recombinant factor VIII concentrates.[54]
Bethesda assay
If an antibody screen is positive, a Bethesda assay (or one of the modified assays with improved sensitivity, such as the Nijmegen-modified Bethesda assay, or the Centers for Disease Control and Prevention [CDC]-modified Nijmegen-Bethesda assay) is performed to measure the amount of inhibitory antibody present in the patient's sample (reported in Bethesda units [BU]).[55][56] The assay is performed by mixing the patient's plasma with a known amount of factor VIII or factor IX. After a 2-hour incubation period at 37°C (98.6°F), the residual factor VIII or factor IX activity is determined. Patients are classified as low responders or high responders on the basis of inhibitory antibody levels, and this classification contributes to treatment decisions.[1] High responders have a lifetime history of titre ≥5 BU; however, it should be noted that overall high responders may have a low titre result (a titre <5 BU) at any particular test without it altering their overall classification.
Inhibitors can also be quantified using the chromogenic substrate Bethesda assay method, which has the advantage of reducing false positivity.[57]
Antenatal diagnosis
Women who are known carriers and are at risk of having a child with congenital haemophilia may seek antenatal diagnosis.[58] Presence of fetal DNA in maternal blood can be detected as early as early as week 4 to 5 gestation, allowing male sex determination. Sex can also be determined by ultrasound beginning week 11 to 13 gestation. DNA-based analysis of the X chromosome to identify a specific genetic mutation may be performed on amniocentesis or chorionic villus samples (CVS). Amniocentesis is usually done after the 15th week of pregnancy, whereas CVS can be done between 10 and 14 weeks. Limb abnormality has been associated with CVS taken before 10 weeks gestation, and the mother should be counselled about the small risk of spontaneous abortion with both CVS and amniocentesis. If the mutation is known, direct mutation analysis can be performed. If the mutation is not already known, gene sequencing can be done to determine whether a mutation is present. Pre-implantation genetic diagnosis on early embryos is also possible.[38][59]
Use of this content is subject to our disclaimer