Aetiology
Cardiomyopathies can be subdivided according to the classification developed by the American Heart Association Working Group.[1]
Primary cardiomyopathies: genetic
Hypertrophic cardiomyopathy
A relatively common condition affecting about 1 in 500 of the general population, across racial groups.[5][6] Hypertrophic cardiomyopathy (HCM) is characterised by the development of a hypertrophied, non-dilated left ventricle (LV) in the absence of another predisposing condition (such as aortic stenosis or hypertension).[Figure caption and citation for the preceding image starts]: Apical hypertrophic cardiomyopathy: 4-chamber echocardiographic view with contrastAhmed I, Smalley SJ, Zhu DWX, et al. Sudden cardiac arrest in apical hypertrophic cardiomyopathy. BMJ Case Reports. 2009;doi:10.1136/bcr.04.2009.17 [Citation ends].
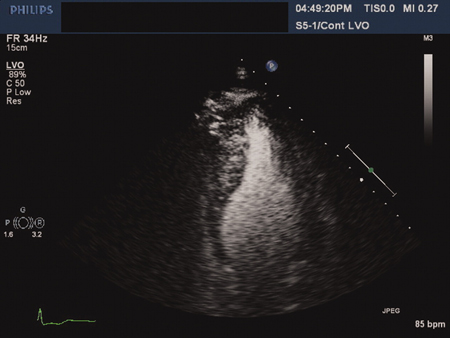 The development of the hypertrophy is characteristically age-related. While early work had suggested that individuals generally developed hypertrophy during adolescence, there is increasing recognition of the development of late-onset disease. In light of this, clinically unaffected adolescent and adult family members should be offered screening in accordance with current guidelines.[7][8][9] European Society of Cardiology (ESC) guidelines provide healthcare professionals with a practical diagnostic and treatment framework for patients of all ages.[7][8] The guidelines also consider the implications of a diagnosis for families.
The development of the hypertrophy is characteristically age-related. While early work had suggested that individuals generally developed hypertrophy during adolescence, there is increasing recognition of the development of late-onset disease. In light of this, clinically unaffected adolescent and adult family members should be offered screening in accordance with current guidelines.[7][8][9] European Society of Cardiology (ESC) guidelines provide healthcare professionals with a practical diagnostic and treatment framework for patients of all ages.[7][8] The guidelines also consider the implications of a diagnosis for families.The hypertrophy is often asymmetrical, but it is important to recognise that the hypertrophy can affect any part of the myocardium. Diastolic dysfunction is often present. Up to one-third of patients have resting left ventricular outflow obstruction, while others may have exercise or other haemodynamic strain-induced outflow gradients.
Atrial arrhythmias are common, and affected individuals have a high risk of thromboembolism.[10]
Approximately 8% of individuals will develop left ventricular systolic dysfunction.[11]
The diagnosis is usually made using a combination of ECG and cardiac imaging, most commonly echocardiography initially.[8][12][13][14] Additional investigations (ambulatory monitoring, exercise stress test) might be appropriate for risk stratification of patients with HCM.[15] These investigations are particularly relevant in individuals with any of the following risk factors: previous cardiac arrest or sustained ventricular tachycardia, non-sustained ventricular tachycardia, extreme left ventricular hypertrophy (>30 mm), unexplained syncope, abnormal blood pressure response to exercise, family history of sudden death, and significant left ventricular outflow tract obstruction.[16][17][18][19]
Cardiac magnetic resonance imaging (MRI) is increasingly being used, as it provides accurate estimation of the extent of hypertrophy, and myocardial fibrosis can be detected as late gadolinium enhancement (LGE).[13][20] Studies have shown that the extent of LGE is suggestive of an increased risk of sudden cardiac death but is not an independent predictor of this. LGE is associated with an increased incidence of non-sustained ventricular tachycardia.[21] US investigators have observed that the presence of late gadolinium may allow for better selection of individuals requiring primary preventative defibrillators.[22] ESC guidelines advise assessing patients with HCM with cardiac MRI and that the presence and extent of LGE may be considered when counselling patients about their risk of sudden death.[8] US guidelines recommend cardiac MRI as a complementary investigation following echocardiography, or as an alternative to echocardiography for those patients in whom the echocardiogram is inconclusive.[9]
An ESC risk prediction model, known as HCM Risk-SCD, has been developed and validated to provide an individualised estimate of 5-year risk of sudden cardiac death.[23] ESC: HCM Risk-SCD Opens in new window
Coronary angiography may need to be undertaken to exclude concomitant coronary artery disease in patients over 40 years of age or who have other coronary risk factors, in particular if they present with typical symptoms of angina or dyspnoea. While myocardial disarray is the pathological hallmark of HCM, endomyocardial biopsy is rarely used as myocardial disarray is often patchy and a small amount of disarray may be found in normal individuals.
The role of genetic testing is beyond the scope of this topic but, traditionally, HCM has been reported to be a sarcomeric disease with mutations reported in genes encoding many sarcomeric proteins, including: beta-myosin heavy chain, myosin binding protein C, troponin T, troponin I, and actin. Other non-sarcomeric genes include PRKAG2 (often associated with pre-excitation) and rarely LAMP-2 (Danon disease).[24] It is important to recognise that there are many other conditions that may present with an HCM phenotype. Such conditions include a range of syndromic conditions such as Fabry disease (particularly in males presenting over the age of 35 years with concentric hypertrophy), Noonan/Leopard syndromes, and Friedreich's ataxia, as well as various other metabolic disorders, mitochondrial diseases, and glycogen storage diseases.[25][26][27] Diagnosis of these conditions requires a high index of clinical suspicion and the use of appropriate diagnostic tools.[28] An important differential diagnosis in young healthy individuals is 'athlete's heart'. Various factors may be used to distinguish HCM from athlete's heart; these include: extent of hypertrophy, LV cavity size, extent of left atrial enlargement, presence of LV outflow tract obstruction, changes in tissue Doppler parameters, peak VO2 consumption, levels of N-terminal-pro brain natriuretic peptide (NT-proBNP), regression on cessation of exercise, and genetic testing.[29]
Arrhythmogenic right ventricular cardiomyopathy (ARVC)
Previously known as arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D).[30] Characterised by progressive replacement of myocardium with fibro-fatty material. These changes are found most commonly in the triangle of dysplasia (right ventricular inflow, outflow, and apex) but are increasingly recognised in the LV. These include clinical variants in which the degree of left ventricular involvement is equal to or greater than the severity of right ventricular involvement, termed biventricular or left-dominant arrhythmogenic cardiomyopathy, respectively.[31]
ARVC represents a wide spectrum of disease, involving both electrophysiological and functional abnormalities. Electrophysiological manifestations range from the most modest of ECG abnormalities to life-threatening ventricular arrhythmias. Similarly, abnormalities of muscle function range from minor contractile changes to severe right and left ventricular failure.[32] Patients present with a wide variety of symptoms ranging from arrhythmias to heart failure to sudden death. In the Veneto region of Italy, ARVC is the most common cause of sudden cardiac death in young individuals.[33]
Diagnosis of ARVC is difficult and requires use of multiple investigations, including: imaging with echocardiography/MRI/angiography, ambulatory monitoring and exercise stress to look for arrhythmias, presence of abnormal resting ECG (T wave abnormalities, delayed conduction in the right-sided leads, epsilon waves), late potential on signal-averaged ECG, and histopathological changes seen on endomyocardial biopsy or at postmortem.
Autosomal dominant inheritance is the most common pattern, although autosomal recessive variants (often with hair and skin changes such as in Naxos disease and Carvajal syndrome) are also seen. Various genes have been implicated - most commonly, desmosomal genes, although there have also been reports of mutations in the RYR2 and TGF-beta genes.[34]
The Task Force criteria for the diagnosis of ARVC/D were published in 1994.[35] However, these criteria have been reported to be too restrictive when dealing with additional family members, so modified criteria were proposed for these individuals.[36] More recently, further modifications of the criteria have been proposed, which include MRI assessment of cardiac structure and give more quantifiable measurements of right ventricular (RV) size and function that are associated with ARVC.[30] There is limited information on the natural history of the disease, although four stages are generally recognised.[37]
Concealed: early, generally asymptomatic phase, although it may present as sudden death.
Overt electrical disorder: unstable phase with symptomatic arrhythmias, generally left bundle branch block, and suggestive of RV origin.
RV failure: phase of progressive deterioration in RV contractile function.
Biventricular pump failure: phase of progressive biventricular dilatation, which may mimic that seen in idiopathic dilated cardiomyopathy (DCM).
Ion channelopathies
Includes long QT syndrome (autosomal dominant or recessive inheritance), Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia (autosomal dominant or recessive forms), short QT syndrome (autosomal dominant inheritance), idiopathic ventricular fibrillation, and sick sinus syndrome (autosomal dominant inheritance). It is important to note that the Working Group of the European Society of Cardiology does not agree with the inclusion of these channelopathies as part of the spectrum of cardiomyopathies.[3]
Other primary genetic cardiomyopathies include conduction system disease and mitochondrial myopathies.
Primary cardiomyopathies: mixed
Dilated cardiomyopathy [Figure caption and citation for the preceding image starts]: Dilated cardiomyopathy: echocardiogramTanejal AK, Wong J, Bayliss J. Antipsychotic-drug-induced dilated cardiomyopathy. BMJ Case Reports. 2009; doi:10.1136/bcr.09.2008.0958 [Citation ends].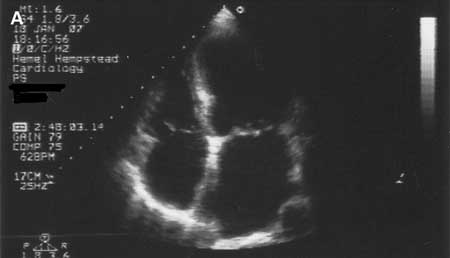
Characterised by the presence of left ventricular dilatation and systolic dysfunction in the absence of abnormal loading conditions or significant coronary artery disease. Right ventricular dilatation is often also present. The prevalence of dilated cardiomyopathy is approximately 1 in 250.[5][6] It is estimated that 30% to 40% of cases are familial; autosomal dominant, autosomal recessive, X-linked, and mitochondrial inheritance patterns have been reported.[6] Familial disease should also be suspected when there is a family history of sudden death, conduction system disease, or an associated skeletal myopathy. Genetic testing of these patients is advised because certain mutations (eg, in the LMNA gene) may indicate a higher risk.[5]
The European Society of Cardiology (ESC) has proposed a revised definition that separates dilated cardiomyopathy into two forms according to imaging parameters: dilated cardiomyopathy and non-dilated left ventricular cardiomyopathy (NDLVC).[8][38]
NDLVC is characterised by non-ischaemic scarring or fatty replacement of the LV in the absence of dilatation and may occur with or without global or regional wall motion abnormalities or isolated LV hypokinesia without scarring that is not explained by abnormal loading conditions or coronary artery disease.[8][38]
Assessment of patients with suspected NDLVC with ECG, echocardiography and cardiac MRI is recommended.
There is significant overlap in the genetic variants implicated in NDLVC with those known to cause both DCM and ARVC.
As this category of cardiomyopathy is relatively novel, little is known about the prevalence and outcomes of this condition. It is felt that genotype strongly influences the risk of sudden death with high-risk genetic variants including; DSP, FLNC, LMNA, DES and RBM20.[8]
Several of the sarcomeric genes that have been implicated in the development of hypertrophic cardiomyopathy may also lead to a dilated cardiomyopathy phenotype.[39] Mutations in the lamin A/C gene may result in a wide variety of different phenotypes, but from a cardiac perspective, diagnosis of a laminopathy is important as affected individuals have a high risk of progressive conduction system disease and sudden death.[40] An extensive list of conditions that may lead to dilated cardiomyopathy has been recognised. One well-known cause is post-myocarditis, which is often asymptomatic from a patient's perspective. Other causes include: alcohol; chemotherapeutic agents (anthracyclines, trastuzumab); storage diseases (e.g., haemochromatosis); autoimmune and systemic disorders; neuromuscular disorders; mitochondrial, metabolic/endocrine (thyroid, diabetes, acromegaly, phaeochromocytoma), and nutritional disorders (in particular, thiamine, selenium, and protein malnutrition problems).[41] If no cause is identified, despite extensive investigation, the term idiopathic dilated cardiomyopathy is applied.
Ischaemic cardiomyopathy
A common consequence of myocardial ischaemia leading to cardiac dysfunction. It is important to note that this is excluded from the classification outlined in the American Heart Association scientific statement.[1]
Restrictive cardiomyopathy
A less well-defined cardiomyopathy as its diagnosis is based on establishing the presence of a restrictive ventricular filling pattern. The European Society of Cardiology Working Group defines it as: 'restrictive physiology in the presence of normal or reduced diastolic volumes, normal or reduced systolic volumes and normal wall thickness'.[3] In many ways, it is a description of the pathophysiological state, which may represent various pathologies or cardiomyopathies rather than a distinct cardiomyopathy.
It may be idiopathic, familial (has been related to troponin I or desmin mutations, the latter often in association with a skeletal myopathy), or associated with various systemic disorders, such as haemochromatosis, amyloidosis, sarcoidosis, Fabry disease, carcinoid syndrome, scleroderma, anthracycline toxicity, or previous radiation. In the paediatric population some rare metabolic syndromes (Gaucher's disease or Hurler's syndrome) must be excluded.
Endocardial pathology (hypereosinophilic syndromes or endomyocardial fibrosis) may also result in a restrictive cardiomyopathy.[42] Patients may present with symptoms of dyspnoea or palpitations, and these relate to diastolic dysfunction. There is usually an elevated jugular venous pressure. The ECG is typically abnormal with evidence of bi-atrial hypertrophy and non-specific ST-T wave changes. Cardiac imaging by echocardiography or MRI shows bi-atrial enlargement with evidence of diastolic dysfunction, evident on mitral inflow Doppler patterns as well as tissue Doppler signals.
As it may present in a similar manner to constrictive pericarditis, restrictive cardiomyopathy must be distinguished from that condition, and this may require cardiac catheterisation. Endomyocardial biopsy may be helpful in selected cases.[43] Diuretics are used for symptomatic control but caution is required as they may reduce preload and thus be counterproductive. Patients may have complex arrhythmias, including atrial fibrillation, and implantable cardioverter-defibrillators (ICDs) may be needed for the management of ventricular tachyarrhythmias.
Primary cardiomyopathies: acquired
Inflammatory myocarditis
An acquired acute or chronic inflammatory condition affecting the myocardium, which can result from a large number of causes, including infectious agents, toxins, and drugs. Three phases are recognised: active, healing, and healed stages. The standard Dallas pathological criteria require that an inflammatory infiltrate with or without myocyte necrosis is present on standard histological assessment.[44] However, newer criteria have been developed that are based on cell-specific immunoperoxidase stains for specific antigens.
There are significant risks associated with endomyocardial biopsy, and this has led to recommendations based on the likelihood of finding specific diseases.[45] Two scenarios that describe the most common presentations of fulminant myocarditis and giant-cell myocarditis are: 1) unexplained new-onset heart failure <2 weeks duration in association with a normal size or dilated LV and haemodynamic compromise; and 2) unexplained new-onset heart failure of 2 weeks to 3 months duration in association with a dilated LV and evidence of ventricular arrhythmias or high-degree atrioventricular block or a failure to respond to usual care within 1 to 2 weeks. Cardiac MRI may provide an alternative diagnostic tool as studies have shown a good correlation between active regions of myocarditis and areas of abnormal signal intensity on MRI.[13]
Takotsubo syndrome
A condition that is manifest by symptoms and signs of acute myocardial infarction in the absence of significant coronary artery disease or spasm. (The condition derives its name from the fact that the heart takes on the appearance of a Japanese octopus fishing pot.)[46] Some authors refer to the condition as 'transient apical ballooning and stress cardiomyopathy'. [Figure caption and citation for the preceding image starts]: Left ventriculogram demonstrating apical ballooning in tako-tsubo cardiomyopathyAugustine DX, Domanski A, Garg A. The stress of chest pain: a case of tako-tsubo cardiomyopathy. BMJ Case Reports. 2009; doi:10.1136/bcr.03.2009.1660 [Citation ends].
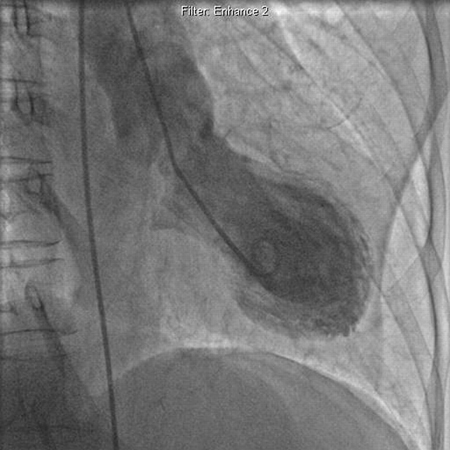
Patients present with chest pain associated with ST elevation, T wave inversion, and mild elevations in cardiac markers. It is most commonly seen in postmenopausal women, and is usually preceded by some form of significant emotional or physical stress.[47] During the acute event, transient elevations in noradrenaline (norepinephrine) levels have been recorded (in the absence of a phaeochromocytoma). If the patient survives the acute event, left ventricular function usually normalises over a period of several weeks.
Various groups have proposed diagnostic criteria. The most widely used are those from a group working at the Mayo Clinic (diagnostic criteria initially reported in 2004 and then modified in 2008):[46]
Transient hypokinesis, akinesis or dyskinesis of the left ventricular midsegments with or without apical involvement; the regional wall abnormalities extend beyond a single epicardial vascular distribution; a stressful trigger is often but not always present
Absence of obstructive coronary disease or angiographic evidence of acute plaque rupture
New ECG abnormalities (ST elevation and/or T wave inversion) or modest elevation in cardiac troponin
Absence of phaeochromocytoma/myocarditis.
Peripartum
Peripartum cardiomyopathy (PPCM), as defined by The Working Group on PPCM of the European Society of Cardiology, is an idiopathic cardiomyopathy frequently presenting with heart failure secondary to LV systolic dysfunction (LVEF <45%), towards the end of pregnancy or in the months following delivery, where no other cause of heart failure is found.[48][49] It is uncommon, occurring in approximately 1 in 4000 births.[50] Reports suggest it is more common in women over the age of 30 and there is an association with gestational hypertension, and twin pregnancy. PPCM appears to be much more common in women of African-American descent with a low incidence in Hispanic women. Some patients present in early pregnancy, and have been classified as having early pregnancy-associated cardiomyopathy.[51]
Several mechanisms (such as myocarditis, immunological, and haemodynamic causes) have been proposed. The condition may be associated with complete recovery in up to 50% of cases, while in others it may progress to severe heart failure resulting in cardiac transplantation or death. It appears to carry significant morbidity.[52] Oxidative stress and the generation of a cardiotoxic subfragment of prolactin may play key roles in the pathophysiology of PPCM. As such, pharmacological blockade of prolactin offers the possibility of a disease-specific therapy.[48]
Tachycardia-induced
In certain individuals, prolonged periods of fast heart rate associated with atrial tachycardia, atrial fibrillation, or ventricular arrhythmias may lead to a tachycardia-related cardiomyopathy.[53] This is an important condition to recognise as the cardiomyopathy may be reversible when rate control has been achieved.[54]
Secondary causes of cardiomyopathy
Secondary cardiomyopathies demonstrate myocardial involvement in the context of generalised, systemic (multi-organ) disorders.[1]
Infiltrative or storage diseases often result in cardiomyopathy, and causes include the following.
Amyloidosis (hypertrophic or restrictive): characterised by the deposition of amyloid fibrils in the heart and includes a wide spectrum of disease, including primary amyloidosis, familial amyloid polyneuropathy, and senile cardiac amyloidosis.[55] Characteristically, patients present with symptoms and signs of heart failure or rhythm disturbances.[56][57] The diagnosis may be suspected when patients have had an ECG or echocardiogram undertaken as part of an initial cardiac workup. The ECG is characterised by the presence of small QRS voltages, a pseudo-infarction pattern in the anterior or inferior leads, and ventricular and supraventricular arrhythmias. The echocardiogram is notable for increased thickening of the atrial septum and left ventricular wall; increased myocardial echogenicity (speckled), associated with some valve thickening; and often a small pericardial effusion. One of the hallmarks of the condition is the inverse relationship between LV mass on cardiac imaging and ECG voltage.[56][58] Increasingly, cardiac MRI is used as a diagnostic tool.[8][13][56][59] MRI reveals a diffuse decrease in the T1 and T2 signal of the myocardium, corresponding with amyloid infiltration, and a characteristic appearance of global sub-endocardial LGE. Biopsy of an affected tissue often provides histological confirmation.[56][58]
Gaucher's disease (restrictive).
Mucopolysaccharidoses (Hunter's syndrome and Hurler's disease; mainly dilated cardiomyopathy).[60]
Haemochromatosis (dilated or restrictive cardiomyopathy): classical haemochromatosis is an autosomal recessive disease associated with a mutation of the HFE gene, which leads to iron overload. It is a multisystem disorder, in which cardiac involvement is manifest by the development of a dilated cardiomyopathy leading to heart failure and conduction system disorder.[57] Treatment with iron depletion may reverse early changes. Cardiac involvement and detection of iron is easily achieved by cardiac MRI.[8][13]
Fabry disease (hypertrophic or restrictive cardiomyopathy).
Pompe's disease (glycogen storage disease type 2; hypertrophic cardiomyopathy).
Niemann-Pick disease (hypertrophic cardiomyopathy).
Iron overload states, including thalassaemia.
Several toxins are associated with the development of cardiomyopathy.
There is a strong correlation between doxorubicin and the development of cardiomyopathy.[61] Cardiomyopathy may also occur as a result of trastuzumab therapy.[62]
Chronic alcohol consumption may damage myocardial tissue directly by inducing apoptosis, or may be associated with thiamine deficiency and the development of wet beriberi (although the exact mechanism is not known).[63]
Cobalt added to beer and exposure to certain chemicals such as heavy metals may also cause myocardial damage.[64]
Endocrine disorders are known to be associated with cardiomyopathy, although evidence is limited to case reports, and the resulting cardiomyopathy can be dilated or restrictive.
Diabetes mellitus may cause an infiltrative/restrictive cardiomyopathy due to glycosylation of the myocardium and subsequent diastolic dysfunction.[65] It is important to exclude associated coronary heart disease in patients with diabetes mellitus who present with a suspected cardiomyopathy.
Cardiomyopathy can also be caused by hyperthyroidism/hypothyroidism, hyperparathyroidism, phaeochromocytoma and acromegaly.
Cardiomyopathies are a recognised feature of certain cardiofacial syndromes.
Noonan syndrome.
Lentiginosis.[26]
Nutritional deficiencies of various vitamins and minerals have been associated with the development of cardiomyopathy.
Thiamine deficiency causes high-output heart failure known as wet beriberi. Many pathophysiological mechanisms have been proposed, but it is thought that initial mitochondrial injury may subsequently lead to cardiomyopathy.[66]
Other nutritional deficiencies involving vitamin C, niacin, selenium, and vitamin D may also cause cardiomyopathy.
Other causes
Neuromuscular and neurological disorders such as Friedreich's ataxia, muscular dystrophies (Duchenne, Becker, and Emery-Dreifuss muscular dystrophy; myotonic dystrophy) can present with cardiac involvement.
Autoimmune disorders such as systemic lupus erythematosus.
Sarcoidosis is a systemic granulomatous disorder of unknown cause. Overt cardiac involvement is uncommon, and patients may present with heart failure (due to development of cardiomyopathy), bradyarrhythmias/high grade atrioventricular block, or ventricular tachyarrhythmias leading to sudden death.[67] Subclinical myocardial involvement is common, and cardiac involvement is often only identified only at autopsy. Granulomatous involvement is often patchy and the yield from endomyocardial biopsy relatively low. Various diagnostic criteria have been developed.[68] Patients with cardiac sarcoidosis and symptoms of syncope should have aggressive investigation and be considered for device-based therapy, including ICD. Cardiac MRI is increasingly being used as a useful imaging technique and FDG-PET scanning can be useful for both the identification of cardiac sarcoidosis and for monitoring disease activity.[13][69][70][71][72] Referral to other specialists may be necessary for immunosuppressive treatment of the underlying condition.
Electrolyte imbalance: commonly potassium, phosphate, or magnesium.
Endomyocardial fibrosis.
Loeffler's endocarditis/hypereosinophilic syndrome.
Use of this content is subject to our disclaimer