Aetiology
The aetiology of most forms of primary aldosteronism (PA) is unknown. The occurrence of PA within families is in keeping with a genetic basis for at least some forms of this condition.[10][41][43] One dominantly inherited and glucocorticoid-remediable variety of PA is familial hyperaldosteronism type I (FH-I). First described in 1966, FH-I is caused by a hybrid gene, which is composed of 11-beta-hydroxylase gene (CYP11B1) sequences at its 5' end and aldosterone synthase gene (CYP11B2) sequences at its 3' end.[44][45][Figure caption and citation for the preceding image starts]: Hybrid CYP11B1/CYP11B2 gene responsible for ACTH-regulated aldosterone overproduction in FH-IFrom the personal collection of Dr Michael Stowasser; used with permission [Citation ends].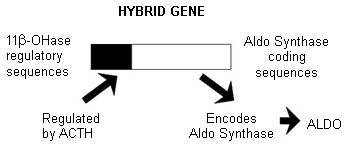
In 1991, a second familial variety of PA, familial hyperaldosteronism type II (FH-II), was described that was neither glucocorticoid-remediable nor associated with the hybrid gene.[12][46] Although the term 'FH-II' was originally used to describe familial forms of PA other than FH-I, this term has been reassigned to familial PA caused by germline mutations within CLCN2 after the largest of the originally described families (and several other probands with PA) was found to have a mutation in that gene.[17] Characteristic clinical features of FH-II include early onset (<20 years of age) hypertension, a relatively mild form of PA in which hypertension is usually readily controlled with medications that block aldosterone action, minimal if any morphological abnormalities of the adrenal on imaging studies, and lack of responsiveness of aldosterone to upright posture.
A family with early and severe PA, markedly elevated levels of 'hybrid steroids' (18-hydroxy- and 18-oxocortisol), and marked zona fasciculata hyperplasia, first reported in 2008, and designated as having familial hyperaldosteronism type III (FH-III), was reported to have a germ-line mutation within KCNJ5 (encodes a potassium channel).[14][15][47] Germ-line KCNJ5 mutations appear to be a rare cause of bilateral PA and are associated with an early onset (<18 years of age) but with variable severity (from mild to very florid PA) that is dependent on the type of mutation inherited.[16]
Germ-line mutations in CACNA1H (encodes a voltage-gated calcium channel) have also been identified in a small number of individuals with onset of PA before 10 years of age.[48] In some of these individuals, the mutation was inherited from a parent, giving rise to the term 'familial hyperaldosteronism type IV' to describe PA resulting from germ-line CACNA1H mutations. Germ-line mutations in CACNA1D (also encodes a voltage-gated calcium channel) were also found in two individuals with early onset PA (but as yet no known affected relatives) who suffered from seizures and other neurological disorders.[49]
Families with PA of uncertain genetic aetiology have been reported to be at least 5 times more common than FH-I.[23] Both aldosterone-producing adenoma and bilateral adrenal hyperplasia (BAH) are represented, often within the same family.[13][22] These patients are clinically, biochemically, and morphologically indistinguishable from those with apparently sporadic PA, and responsible mutations may therefore underlie the development of PA in other patients who lack a family history of this condition.[4][13][22][23]
Somatic mutations in KCNJ5 were identified in 8 of 22 large, apparently sporadic aldosterone-producing adenomas.[47] Other groups have reported somatic KCNJ5 mutations to be present in 30% to 40% of aldosterone-producing adenomas removed from white patients and in >60% removed from patients from Japan and China.[50][51][52] Somatic mutations have since been identified in ATP1A1 (encodes the α-subunit of Na+/K+ ATPase), ATP2B3 (a Ca2+ ATPase calcium channel), and CACNA1D (encodes a voltage-gated calcium channel) in much smaller proportions (5%, 2%, and 11% respectively) of APAs.[49][53][54]
Studies involving immunohistochemical techniques have revealed the presence of abnormal foci of CYP11B2-expressing cells, termed aldosterone-producing cell clusters (APCCs), within adrenal cortices, which become more numerous with advancing age.[55] Concurrently, zona glomerulosa, typically continuous in childhood and early adulthood, becomes increasingly disrupted and relatively suppressed in terms of CYP11B2 expression. These observations support a potential pathophysiological mechanism (APCC formation) underlying the apparent gradual development of autonomous aldosterone production with ageing (as evidenced by rising ARR), and which may predispose to the development of frank PA, due either to bilateral adrenal hyperplasia (BAH) or to aldosterone-producing adenoma (APA) (or both).
Pathophysiology
In all forms of PA, aldosterone production is excessive to the body's requirements and relatively autonomous with regard to its normal chronic regulator, the renin-angiotensin II system.[56] This results in excessive sodium re-absorption through amiloride-sensitive epithelial sodium channels within the distal nephron, leading to hypertension and suppression of renin-angiotensin II. Urinary loss of potassium and hydrogen ions, exchanged for sodium at the distal nephron, may result in hypokalaemia and metabolic alkalosis if severe and prolonged enough. The exact causes of excessive, autonomous aldosterone production in aldosterone-producing adenoma and bilateral adrenal hyperplasia are unknown, but genetic factors related to adrenal cortical cellular growth regulation and/or steroid biosynthesis are likely to be involved. Abnormal foci of CYP11B2-expressing cells, termed APCCs, have been detected within adrenal cortices and become more numerous with advancing age.[55] Concurrently, renin becomes progressively suppressed, whereas aldosterone does not but becomes less responsive to salt, suggesting that aldosterone production by these APCCs may be constitutive and not responsive to renin. The clinical significance of these APCCs is uncertain, and includes the possibilities that they represent a pathological basis for BAH, precursors of APAs, or a new form of PA altogether.[57]
In FH-I, the causative hybrid gene encodes a hybrid enzyme of unique structure that synthesises aldosterone but, unlike CYP11B2, is regulated by adrenocorticotrophic hormone (ACTH) and not by angiotensin II.[45] Aldosterone production in FH-I is therefore regulated by ACTH rather than by angiotensin II, and can be suppressed and managed by administering small doses of glucocorticoids such as dexamethasone.[44]
Mutations in KCNJ5 (which encodes an inwardly-rectifying potassium channel) lead to reduced potassium/sodium channel selectivity and sodium influx, predisposing to cell membrane depolarisation, increased calcium influx, increased expression of genes promoting aldosterone synthesis, and increased aldosterone production by adrenocortical cells.[16][47] Mutations in ATP1A1, ATP2B3, CACNA1H, and CACNA1D also appear to share increased calcium influx as a common pathophysiological mechanism for increased and autonomous production of aldosterone.[49][53][54] Mutations in CLCN2 predispose to cell membrane depolarisation and possibly also to increased calcium influx.[17] How these effects lead to adrenal cell proliferation and tumour development remains uncertain.
Although morbidity in PA mainly results from hypertension, experimental, and clinical evidence strongly suggests that aldosterone excess can bring about adverse cardiovascular sequelae (including remodelling and fibrosis) independently of its hypertensive effects.[58][59] In animal studies, both aldosterone excess and a high salt intake appear to be necessary for induction of cardiac fibrosis,[58] and coronary vasculitis has been observed to be an early manifestation.[60] These effects were preventable by the administration of mineralocorticoid receptor antagonists.[58][60] The doses of aldosterone used in experimental studies have been very large, and the results of these studies may, therefore, have limited applicability to clinical situations. Nevertheless, several groups have convincingly demonstrated abnormalities in cardiovascular morphology or function in patients with PA that appear to be out of proportion to the elevation in BP.[59][61][62][63][64] These have included:
Increased left ventricular mass index and reduced diastolic function, both of which markedly improved following specific treatment of PA[59][61]
Reduced myocardial perfusion at rest and during exercise[62][63]
Increased myocardial backscatter (an echo marker of myocardial fibrosis)[64]
Increased proteinuria (as evidence of renal glomerular damage)[65]
A greater incidence of cardiovascular events, which was reversed following specific surgical or medical treatment.[66][67]
Evidence of left ventricular remodelling was also reported in individuals with genetically proven FH-I who had biochemical evidence of aldosterone excess but had not yet developed hypertension.[68]
Classification
Pathological classification[3][4][5]
Aldosterone-producing adenoma (APA): a benign adrenocortical tumour of at least 10 mm in diameter autonomously producing aldosterone.[Figure caption and citation for the preceding image starts]: Aldosterone-producing adenomaFrom the personal collection of Dr Michael Stowasser; used with permission [Citation ends].
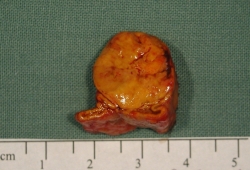 APAs may be further sub-classified according to whether they are angiotensin-unresponsive (as in the classic Conn’s tumour) or angiotensin-responsive, in which responsiveness is defined as a rise in plasma aldosterone by at least 50% over basal during 2 or 3 hours of upright posture following overnight recumbency or during an infusion of angiotensin II.
APAs may be further sub-classified according to whether they are angiotensin-unresponsive (as in the classic Conn’s tumour) or angiotensin-responsive, in which responsiveness is defined as a rise in plasma aldosterone by at least 50% over basal during 2 or 3 hours of upright posture following overnight recumbency or during an infusion of angiotensin II.Aldosterone-producing nodule (APN): a benign adrenocortical lesion of less than 10 mm in diameter autonomously producing aldosterone.
Aldosterone-producing adrenocortical carcinoma (APACC): a malignant adrenocortical tumour autonomously producing aldosterone.
Other unilateral forms: one adrenal excessively and autonomously producing aldosterone, but with no discrete tumour identified on pathological examination. Instead, the adrenal is shown to contain either one or multiple aldosterone-producing micronodules (APM or MAPM) that stain positive for CYP11B2 by immunohistochemistry but are not distinguishable from the surrounding cortex by haematoxylin-eosin staining. More rarely, aldosterone-producing diffuse hyperplasia (APDH) shows a broad continuous region of hyperplastic, CYP11B2-positive zona glomerulosa cells.
Bilateral forms: both adrenals affected by diffuse and/or nodular hyperplasia and excessively and autonomously producing aldosterone; includes both non-glucocorticoid-remediable (idiopathic) and glucocorticoid-remediable forms. Rarely, bilateral hyperplasia is macronodular with autonomous secretion of cortisol more often than of aldosterone.
Functional (treatment-oriented) classification[6][7][8][9][10]
Unilateral PA:
Includes APA, APN, APACC, and unilateral (or primary) adrenal hyperplasia.
Bilateral PA:
Non-glucocorticoid-remediable: includes bilateral (idiopathic) adrenal hyperplasia which is rarely macronodular (non-familial, familial hyperaldosteronism type II, and familial hyperaldosteronism type III) and bilateral APA.
Glucocorticoid-remediable (familial hyperaldosteronism type I).
Familial classification[4][10][11][12][13][14][15][16][17]
Familial hyperaldosteronism type I (FH-I; glucocorticoid-remediable; associated with hybrid gene).
Familial hyperaldosteronism type II (FH-II; non-glucocorticoid-remediable; associated with germ-line mutations of CLCN2).
Familial hyperaldosteronism type III (FH-III; non-glucocorticoid-remediable; associated with germ-line mutations of KCNJ5).
Familial hyperaldosteronism type IV (FH-IV; non-glucocorticoid-remediable; associated with germ-line mutations of CACNA1H).
Apparently non-familial PA (without known affected relatives).
Use of this content is subject to our disclaimer