Aetiology
The aetiology of Kawasaki disease (KD) remains unknown. However, the following observations suggest that this disease is triggered by an unknown infectious agent in a susceptible host.
Seasonal variation: variation in incidence associated with different seasons with peaks noted in Japan and the US in the winter and spring.[11]
Geographical distribution: temporal clusters have been reported in the US, Japan, and worldwide.[3] Moreover, in Japan, hot spots of increased incidence have been noted to cluster in certain geographical areas.[13]
Age at onset: peak incidence is in early childhood, with 80% cases in infants aged <5 years. The rarity of cases in infants aged <3 months may suggest protective transplacental antibodies.[3][9]
To date, no infectious trigger has been identified for KD with studies specifically failing to find any link with parvovirus B19, retrovirus, Epstein-Barr virus, herpes, measles, or human coronavirus (NL-63).[14]
Similar phenotypes of toxin-mediated diseases (including staphylococcal and streptococcal toxic shock syndrome [TSS] and scarlet fever), with fever, mucosal membrane involvement and desquamating skin rash, have also led to certain bacteria being considered as putative triggers for KD. Moreover, TSS toxin-secreting Staphylococcus aureus isolated from a patient with KD, manifested with coronary aneurysm, has been reported.[15] It is speculated that a trigger, potentially an infection, results in an immune-mediated reaction, leading to disease manifestation in an immunogenetically susceptible host. It has been proposed that, in both KD and TSS, the disease is caused by viral or bacterial toxins acting as superantigens.[16][17] A superantigen theory for KD continues to be investigated; however, significant supportive data are currently lacking.[18]
The genetics of KD are complex and it is likely that multiple polymorphisms contribute to the pathogenesis. Genetics are thought to play an important role given the increased incidence of KD in Asian populations in the US and other geographical areas. In addition, there is familial aggregation with increased incidence in parents and siblings of affected children.[11] In the largest KD genome-wide association study in Japan, the most significantly associated genetic susceptibilities were variants associated with high affinity of FC receptor for immunoglobulin G (FCGR2A) and variants related to the T-cell receptor region regulator known as ITPKC (inositol 1,4,5-triphosphate 3-kinase C).[19][20] Variants in the latter are associated with coronary artery aneurysm developement and intravenous immunoglobulin resistance.[19]
One 2017 meta-analysis of genetic association studies found 23 gene polymorphisms to be associated with KD susceptibility, and 10 that may be associated with coronary artery lesions. Many of these genes are lymphocyte activators or inhibitors with others involved in cytokine, chemokine, and adhesion molecule function, or vascular remodelling.[21]
Although similarities exist between KD and acrodynia (mercury hypersensitivity), studies to link KD to drugs, toxins, chemicals, and heavy metals have shown negative results.[22]
Pathophysiology
Initially, KD was considered a self-limiting, benign condition, however, it is now known that 20% to 25% of untreated patients develop coronary artery aneurysms, and there is an up to 2% mortality rate associated with coronary artery aneurysms. In addition, KD has been reported to be associated with myocardial infarction, sudden death, and ischaemic heart disease.[23]
In the early phase of the disease, there is development of oedema and neutrophil infiltration in the coronary arterial wall, with a rapid transition to mononuclear cells.[24] This is followed by local production of matrix metalloproteinases that cause destruction of the internal elastic lamina and media. Intimal myofibroblastic proliferation causes fibrous connective tissue replacement of the intima and media, leading to aneurysm formation, scarring, and stenosis.[25] Fibrotic changes have been identified of the myocardium, emphasising that the cardiac inflammation may be more widespread than initially considered, and not restricted to the coronary arteries.[12]
Arterial remodelling, a process which can take place over years, can lead to stenoses, particular at the interface between the normal artery and the aneurysm. Larger aneurysms are more likely to persist and are associated with an increased risk of thrombotic coronary occlusion, stenoses and myocardial infarction.
Reports are available from children with KD who did not develop coronary abnormalities during the acute phase of the disease and died years later due to unrelated causes. Autopsies performed on these children demonstrated coronary artery intimal thickening and medial fibrosis.[26] It is uncertain if such cases represent late sequelae of missed aneurysms following the acute presentation; or more worringly an emergence of late KD coronary artery vasculopathy independent of the presence of coronary artery aneurysms following the acute disease.
Classification
Clinical stages[1]
Clinically, the course of untreated KD is divided into the following stages:
Acute febrile stage (lasting weeks 1-2)
Fever, irritability, cervical adenitis, conjunctivitis, rash, mucosal erythema, painful erythema of the hands and feet, arthralgia or arthritis, possible myocarditis, and pericarditis.
Subacute stage (lasting weeks 2-4)
Fever, rash, and lymphadenopathy have resolved; if fever persists there is an increased risk of cardiac complications; persistent irritability, poor appetite, and conjunctival injection; desquamation of extremities begins at this stage.
The patient may be completely asymptomatic if given intravenous immunoglobulin (IVIG). Periungual desquamation may be the only apparent clinical manifestation.
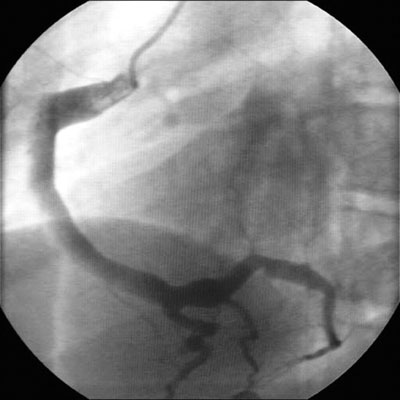
Cardiac abnormalities (coronary artery ectasia or aneurysms) may develop during this stage, and rarely, later in patients treated with IVIG.[Figure caption and citation for the preceding image starts]: Coronary artery ectasiaBMJ Case Reports 2009; doi:10.1136/bcr.10.2008.1113 [Citation ends].
Convalescent (lasting weeks 4-8)
All signs of inflammation have receded, and acute phase markers normalise.
If present, coronary artery ectasia or aneurysms may persist and enlarge.
Chronic stage (variable)
If present, coronary artery dilation may resolve.
However, coronary artery aneurysms may persist through adulthood. Such patients are at risk of subsequent coronary artery thrombosis, rupture, and myocardial infarction.
Use of this content is subject to our disclaimer