Approach
Pseudohypoparathyroidism (PHP) is a rare condition, and referral to a specialised centre should be considered. One 2018 international consensus statement and one 2020 review provides recommendations for diagnosis and management of PHP.[1][2] Patients may be asymptomatic or present with symptoms and signs of hypocalcaemia. In addition, patients with types 1a and 1c PHP have the characteristic skeletal phenotype of Albright's hereditary osteodystrophy and often present with short stature. Subcutaneous calcification has been reported in the neonatal period.[29] A proportion of patients may have concurrent hypothyroidism. The different forms of PHP are distinguished by using a combination of clinical features, measurement of the response of urinary cyclic adenosine monophosphate (cAMP) and phosphate to exogenous parathyroid hormone (PTH) stimulation, and genetic testing.
Types 1a and 1c PHP have an autosomal dominant mode of inheritance.[26] Type 1b is usually sporadic, although familial cases have been reported.[27][28]
The inheritance of type 2 PHP is unknown, but it has been hypothesised that type 2 PHP may be an acquired defect secondary to vitamin D deficiency.[6]
PTH resistance
PTH resistance produces hypocalcaemia and hyperphosphataemia, which produce distinct clinical features. Hypocalcaemia does not always manifest at birth but tends to occur between 3 and 8 years of age. Mild hypocalcaemia is often asymptomatic. Some patients may report muscle cramps. Severe hypocalcaemia is a medical emergency, and it is important to be alert for the key symptoms and signs. Paraesthesias affecting the lips, fingers, or toes are common early symptoms. Severe hypocalcaemia can cause muscle twitches, spasm, or tetany. If spasms affect laryngeal smooth muscle, the patient may develop life-threatening stridor. The patient may report lethargy or anxiety. If left untreated, patients can develop confusion, convulsions, or a fatal cardiac arrhythmia. PHP may also be a cause of paroxysmal dyskinesias (involuntary intermittent movement disorders).[30] Subtle signs such as brittle nails, dry hair, or calcification of the pinna of the ears may also be seen.
Hypocalcaemia produces two key clinical signs.
Chvostek's sign is elicited by tapping a finger on the facial nerve in front of the tragus of the ear with the mouth slightly opened. Twitching of the ipsilateral facial muscles is considered positive and is a sign of hypersensitivity of the nerve fibres.
Trousseau's sign is elicited by inflating a blood pressure cuff placed over the brachial artery to 20 mmHg above systolic for 5 minutes. The distal ischaemia produces tetany of the hand with flexion at the metacarpophalangeal joints and extension at the interphalangeal joints. The more rapid the response, the lower the serum calcium.
Hyperphosphataemia produces an increase in the deposition of calcium phosphate in extraskeletal tissues. Calcium phosphate stones can form in the kidney, producing nephrolithiasis. Calcium phosphate deposition in the brain produces basal ganglia calcification, a classic computed tomographic (CT) finding of PHP. Calcium phosphate deposition in the skin produces subdermal calcification and ossification. Calcium phosphate deposition in the eye can cause cataracts. Routine ophthalmological examination is required in all patients to identify cataract formation.
[Figure caption and citation for the preceding image starts]: Eliciting Chvostek's signCooper MS, Gittoes NJL. BMJ. 2008 Jun 7;336(7656):1298-302 [Citation ends].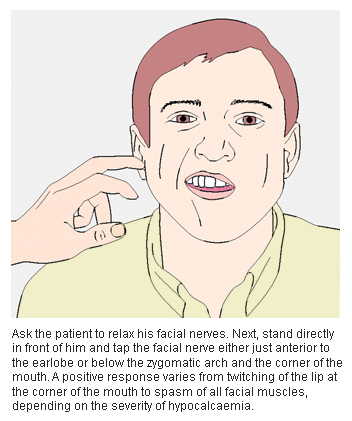 [Figure caption and citation for the preceding image starts]: Eliciting Trousseau's signCooper MS, Gittoes NJL. BMJ. 2008 Jun 7;336(7656):1298-302 [Citation ends].
[Figure caption and citation for the preceding image starts]: Eliciting Trousseau's signCooper MS, Gittoes NJL. BMJ. 2008 Jun 7;336(7656):1298-302 [Citation ends].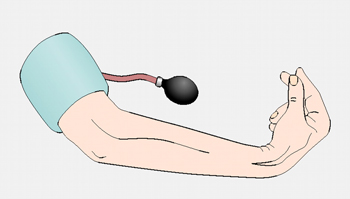
Albright's hereditary osteodystrophy
PHP types 1a and 1c are associated with Albright's hereditary osteodystrophy, which has a characteristic skeletal phenotype. These characteristics include short stature (height <10ᵗʰ percentile), stocky habitus, round face, dental hypoplasia, brachymetacarpals, and brachymetatarsals. Most patients have associated intellectual disability and obesity. The most useful diagnostic sign is metacarpal shortening, classically affecting III, IV, and V metacarpals, which produces dimpling over the knuckles.[31]
Other congenital disorders may have a similar phenotype and can be difficult to distinguish.[32] However, these patients do not have any demonstrable defects in PTH signalling or function.[Figure caption and citation for the preceding image starts]: Child with Albright's hereditary osteodystrophy showing a round face and a short nose with a flat nasal bridgeWilson LC, Trembath RC. J Med Genet. 1994 Oct;31(10):779-84 [Citation ends].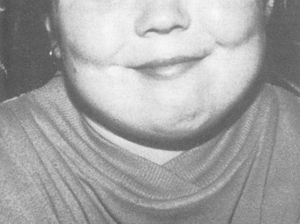 [Figure caption and citation for the preceding image starts]: Hands of an adult with Albright's hereditary osteodystrophy showing shortening of the IV metacarpal and distal phalanges, and knuckle dimples in the clenched fistsWilson LC, Trembath RC. J Med Genet. 1994 Oct;31(10):779-84 [Citation ends].
[Figure caption and citation for the preceding image starts]: Hands of an adult with Albright's hereditary osteodystrophy showing shortening of the IV metacarpal and distal phalanges, and knuckle dimples in the clenched fistsWilson LC, Trembath RC. J Med Genet. 1994 Oct;31(10):779-84 [Citation ends].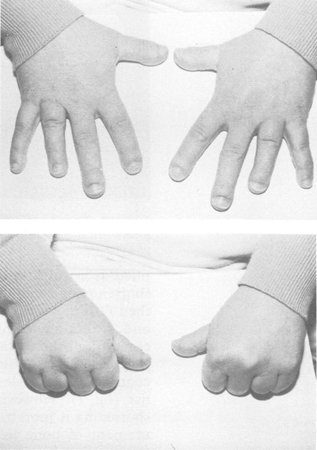
Other endocrine abnormalities and associations
The most common associated endocrine dysfunction is hypothyroidism, produced by decreased responsiveness to thyroid-stimulating hormone (TSH). The clinical features of hypothyroidism are non-specific and include weakness, lethargy, cold sensitivity, constipation, weight gain, depression, menstrual irregularity, myalgia, dry or coarse skin, eyelid oedema, thick tongue, coarse hair, facial oedema, and bradycardia. Hypothyroidism can be seen in any patient with type 1a or 1c disease, and possibly in type 1b disease as well.
Other endocrine dysfunctions are rare and are seen in type 1a or 1c disease. These include gonadotrophin and possibly growth hormone-releasing hormone (GHRH) resistance. Gonadotrophin resistance usually manifests with delayed puberty or infertility. GHRH resistance manifests with features of growth hormone deficiency.
Sporadic cases of Chiari type 1 anomaly, cholesterol gallstones, psychosis, and osteosclerosis have been reported.[33][34][35][36]
Initial tests
Serum calcium (total and ionised), phosphate, and PTH levels should be measured in all patients in whom the diagnosis is suspected. A serum creatinine is also required to aid in the interpretation of calcium and phosphate levels. Hypocalcaemia and hyperphosphataemia associated with elevated PTH concentration and normal 25-hydroxyvitamin D levels strongly suggest a diagnosis of PHP. However, some patients may have normal calcium, phosphate, and PTH levels.
Other tests are performed to exclude other common causes of hypocalcaemia. Serum magnesium levels should be measured to exclude hypomagnesaemia, which can reduce PTH secretion and action. Levels of 25-hydroxyvitamin D should be measured to exclude vitamin D deficiency and osteomalacia.
Subsequent tests
Imaging
In patients with the characteristic skeletal phenotype of Albright's hereditary osteodystrophy, plain radiographs of the hands will reveal metacarpal shortening.[37]
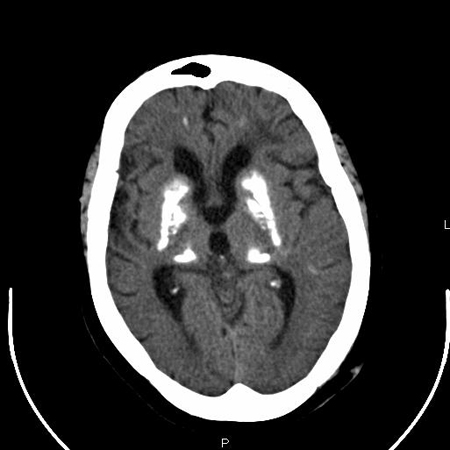
CT scanning of the brain may reveal calcification of the basal ganglia. It is not required for diagnosis, and is usually performed to exclude other causes of paraesthesias and confusion.
Dual-energy x-ray absorptiometry (DXA) bone-density scanning is required in patients with fractures. Bone loss can occur in any form of PHP due to preserved sensitivity of bone to PTH, but it is usually only significant in patients with low sex steroid levels as a result of associated gonadotrophin resistance. Some researchers have suggested that in patients with PHP type 1a, regional bone density is normal while total bone density is elevated.[38]
Response to exogenous PTH
The response to exogenous human PTH (hPTH) is evaluated to distinguish the different subtypes of PHP. It is also the only way to establish the diagnosis of PHP in patients with inconclusive results on initial testing.
The patient is taken off calcium supplements and fasted overnight. At 6 a.m., the patient begins to drink 250 mL fluid every hour until 12 noon. Two control urine specimens for measurement of cAMP, creatinine, phosphate, and calcium are collected before 9 a.m. Subcutaneous synthetic hPTH is given at 9 a.m., and urine is collected every half an hour for measurement of urinary cAMP and phosphorus. Serum samples are collected at 9 a.m. and 11 a.m. for measurement of creatinine, cAMP, 1,25-dihydroxyvitamin D, and phosphorus. The test is concluded at 12 noon.
Normal subjects show a 10- to 20-fold increase in urinary cAMP excretion and a 20% to 30% increase in phosphate excretion regardless of the serum calcium concentration. The cAMP and phosphate response are both decreased in patients with type 1a, 1b, or 1c PHP. Patients with type 2 PHP have a decreased phosphate response, but the cAMP response is preserved. Patients with pseudo-pseudohypoparathyroidism will have a normal cAMP and phosphate response.
Genetic testing
Mutational analysis of the GNAS gene (the gene that codes for the guanine nucleotide-binding protein Gs-alpha) is available through several clinical laboratories. This will reveal known causative mutations of type 1a, type 1b, and pseudo-pseudohypoparathyroidism.[4][39][40][41][42][43] The causes of type 1c and type 2 are unknown.
Other endocrine tests
Type 1a or 1c can have generalised hormone resistance; thyroid function tests (free thyroxine, TSH) should be performed early because thyroid dysfunction is the most common associated endocrine dysfunction seen in these patients.
Evaluation of the gonadotrophin axis (follicle-stimulating hormone, luteinising hormone, oestradiol, or testosterone) and the GHRH-growth hormone axis (growth hormone, insulin-like growth factor-1) should also be considered in all patients with suspected types 1a and 1c PHP, or who have symptoms of delayed puberty or infertility.[Figure caption and citation for the preceding image starts]: CT findings of severe basal ganglia calcification in a patient with pseudohypoparathyroidismFrom the collection of Kent Wehmeier, University of Florida Jacksonville [Citation ends].
Distinguishing PHP clinical subtypes
Using a combination of clinical features, hormone resistance, diagnostic tests, and genetic screening, the different subtypes of PHP can be distinguished.[3][4][6]
[Figure caption and citation for the preceding image starts]: Subtypes of pseudohypoparathyroidismCreated by Dr Emad Naem [Citation ends].
Use of this content is subject to our disclaimer