History and exam
Key diagnostic factors
common
presence of risk factors
Strong risk factors include a family history of delayed puberty (may be specific to constitutional delay or to organic hypogonadotrophic hypogonadism), congenital pituitary abnormalities, known gene mutations, chromosomal disorders, a syndromic diagnosis, anosmia, eating disorders, chronic systemic illness, malnutrition, intense exercise, and congenital and acquired gonadal abnormalities.
boys: testes <4 mL
Testicular size is documented as a measurement of the longest axis or by the testicular volume using the Prader orchidometer.
[Figure caption and citation for the preceding image starts]: Prader orchidometerCreated by BMJ Knowledge Centre [Citation ends]. [Figure caption and citation for the preceding image starts]: Method of comparing testicular size using the Prader orchidometerFrom the collection of Dr A. Mehta [Citation ends].
[Figure caption and citation for the preceding image starts]: Method of comparing testicular size using the Prader orchidometerFrom the collection of Dr A. Mehta [Citation ends].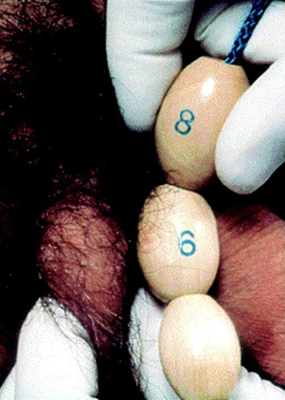 Volume of 4 mL or a longitudinal length of 2.5 cm defines the onset of puberty.[39]f
Volume of 4 mL or a longitudinal length of 2.5 cm defines the onset of puberty.[39]f
girls: absent breast development
The first demonstrable sign of puberty in girls is breast development.[38]
absent pubic/axillary hair
Pubic and axillary hair, acne, and body odour develop as a result of androgens secreted from the adrenal gland. The onset of axillary hair occurs in mid-puberty.
absence of menarche >3 years from breast budding
Menarche serves as well-defined and documented evidence of puberty in girls.
Occurs typically alongside Tanner stage 4 breast development in the majority of girls.
absent growth spurt
The sex hormones directly stimulate the growth plate, resulting in the growth spurt. There is also an increase in growth hormone (GH) secretion.[33] Oestrogen, either from the ovary or aromatised from testicular testosterone, is the factor that mediates the increased GH response during puberty.[34]
The growth spurt occurs in mid- to late puberty in boys and in stage 3 breast development in girls.
On average, it contributes to 25 cm of height in females and 30 cm in males.
anosmia
Kallmann's syndrome is an association of organic hypogonadotrophic hypogonadism and hypoplastic olfactory nerves, resulting in anosmia.[21] Other key signs associated with organic hypogonadotrophic hypogonadism (and known as ‘red flags’ for the condition) include bilateral cryptorchidism, micropenis (2 or more standard deviations smaller than mean length for age), midline defects such as cleft lip, cleft palate, and renal agenesis, as well as synkinesis (mirror movements) that is pathognomonic for this condition.
Other diagnostic factors
common
short stature
Constitutional delay and Turner's syndrome are associated with short stature.
uncommon
dysmorphic features
Girls with Turner's syndrome are short, with several dysmorphic features that include a low posterior hairline, a webbed neck, and prominent posteriorly rotated ears. Presenting features may be more subtle, and investigations to rule out Turner's syndrome should be undertaken in any girl with a delay in puberty.[40]
Patients should be assessed for body disproportion. Boys with Klinefelter's syndrome have tall stature with a greater lower limb length. In addition, they have dysgenetic testes and developmental delay, and they may have gynaecomastia.[27]
There may be other features to suggest a syndromic diagnosis (e.g., Prader-Willi, Bardet-Biedl, CHARGE, or septo-optic dysplasia).
Risk factors
strong
family history of delayed puberty
Constitutional delay in puberty, the most common cause of delayed puberty in both males and females, clusters in families and most commonly displays an autosomal dominant pattern of inheritance.[1][19][20] Between 50% and 75% of individuals with constitutional delay have a family history of delayed puberty.[1]
There is a similarity in the age at puberty between girls and their mothers, especially with age of menarche.
congenital pituitary structural abnormalities
May be due to genetic mutations or to structural abnormalities within the hypothalamo-pituitary axis associated with midline forebrain defects (e.g., septo-optic dysplasia and holoprosencephaly).[22] The resulting gonadotrophin deficiency may be isolated or combined with other pituitary hormone deficiencies.
gene mutations
Several genes have been linked to the pathogenesis of hypogonadotrophic hypogonadism, such as ANOS1, FGFR1, KISS1R, KISS-1, GNRHR, PROK2, PROKR2, NSMF, NROB1, LH and FSH beta-subunit, leptin, leptin receptor, and prohormone convertase 1 (PC1) genes.[24]
chromosomal disorders
Klinefelter's syndrome (XXY), Turner's syndrome (45X), XY gonadal dysgenesis, and 45X/46XY mixed gonadal dysgenesis are associated with dysgenetic gonads and hypergonadotrophic hypogonadism.[27] Males with Klinefelter's syndrome commonly enter puberty but fail to progress fully through puberty, with rising follicle-stimulating hormone concentrations and falling testosterone concentrations from mid-puberty.
syndromic diagnosis
Prader-Willi, Bardet-Biedl, and CHARGE syndromes are associated with hypogonadotrophic hypogonadism.
restrictive eating
Anorexia nervosa and abnormal eating patterns can result in hypogonadotrophic hypogonadism.
chronic systemic illness
Common conditions including chronic heart disease, moderate to severe asthma, cystic fibrosis, coeliac disease, inflammatory bowel disease (Crohn's disease and ulcerative colitis), inflammatory disorders (e.g., juvenile idiopathic arthritis), chronic renal failure, any chronic malignancy, and poorly controlled diabetes mellitus can result in functional hypogonadotrophic hypogonadism.
malnutrition
Can result in functional hypogonadotrophic hypogonadism.
intense exercise
Intense physical exercise can result in delayed onset of puberty, delayed menarche, or arrest in pubertal development.
congenital testicular abnormalities
Hypergonadotrophic hypogonadism can be due to anorchia or testicular regression in males. Severe bilateral cryptorchidism (undescended testes) can also be associated with congenital testicular abnormalities.
acquired gonadal abnormalities
Hypergonadotrophic hypogonadism may result from testicular torsion or tumour.
Autoimmune endocrinopathy can result in gonadal damage and hypergonadotrophic hypogonadism.[29]
pituitary surgery
May result (e.g., following removal of a craniopharyngioma or an adenoma) in hypogonadotrophic hypogonadism.[25]
adrenal hypoplasia
NROB1 mutations cause hypogonadotrophic hypogonadism and adrenal hypoplasia congenita that can result in severe neonatal adrenal crisis. Other mutations implicated in adrenal hypoplasia congenita include those in STAR, CYP11A1, and CYP17A1.[35] The condition is inherited as an X-linked disorder.[23]
weak
chemotherapy
May result in hypergonadotrophic or hypogonadotrophic hypogonadism depending on the chemotherapeutic agent used. Risk for chemotherapy is greatest with alkylating agents.[36]
radiotherapy
May result in hypogonadotrophic hypogonadism with brain/pituitary radiation or hypergonadotrophic hypogonadism following radiation to the pelvis.[30]
histiocytosis
Can result in permanent hypogonadotrophic hypogonadism.
sickle cell disease
Can result in permanent hypogonadotrophic hypogonadism.
iron overload (associated with transfusion)
Can result in permanent hypogonadotrophic hypogonadism.[26]
Use of this content is subject to our disclaimer