Detalhes
As proteínas do complemento moderam as ações de anticorpos específicos, auxiliam no processamento e remoção de imunocomplexos e modificam as respostas de células T e B.
As deficiências do complemento podem ser hereditárias ou adquiridas. As deficiências do complemento adquiridas podem ocorrer como resultado de infecção (por exemplo, infecção meningocócica recorrente ou infecção gonocócica disseminada) ou em conjunto com doença reumatológica ou autoimune crônica (por exemplo, lúpus eritematoso sistêmico ou crioglobulinemia).
O diagnóstico baseia-se na história clínica e/ou familiar e nos achados sorológicos e moleculares característicos; apenas alguns laboratórios especializados fornecem diagnósticos abrangentes.
Apresentações suspeitas incluem meningite meningocócica em indivíduos com idade superior a 5 anos, infecções bacterianas recorrentes, angioedema sem urticária, distúrbios inflamatórios do sistema renal e oftálmico e manifestações autoimunes.
O complemento desempenha uma função importante na defesa contra hospedeiros microbianos. Ele complementa as ações de anticorpos específicos e auxilia no processamento e na remoção de imunocomplexos.[1][2][3][4] Também modifica as respostas das células T e B, empregando receptores específicos encontrados em várias células do sistema imunológico para modular a resposta imune adaptativa.[4] O sistema complemento participa da hematopoiese, do metabolismo lipídico, da reprodução e da regeneração tecidual.[3] Múltiplas interações conectam o complemento com os sistemas de coagulação, fibrinólise e cinina.[5] Devido ao seu poderoso potencial inflamatório, múltiplas proteínas reguladoras são necessárias para prevenir potenciais danos aos tecidos.
Quando hiperativo, o sistema complemento pode causar várias condições inflamatórias e de risco de vida, incluindo sepse, síndrome da resposta inflamatória sistêmica, síndrome do desconforto respiratório agudo e insuficiência de múltiplos órgãos após trauma grave, queimaduras ou infecções.[6] Está implicado em nefropatias graves, bem como em distúrbios neurodegenerativos, como a doença de Alzheimer, a síndrome de Guillain-Barré e a esclerose múltipla.[7][8][9][10] Um sistema complemento ativado em excesso também foi reconhecido como um mecanismo efetor significativo da lesão de reperfusão. A disfunção orgânica pode ser causada pela resposta inflamatória induzida por superfícies artificiais na hemodiálise e nos circuitos extracorpóreos.[11] Nesses casos, isso pode levar a neutropenia transitória, leucostase vascular pulmonar e, mais raramente, choque anafilático.
Estados patológicos ocorrem com deficiências em proteínas do complemento ou defeitos de fatores que controlam, concentram e limitam a ativação do complemento.[12][13][14][15][16] Deficiências do complemento podem ser hereditárias ou adquiridas (secundárias a um estado patológico consumidor do complemento). Deficiências do complemento podem elevar o risco de infecção bacteriana invasiva e/ou estar associadas com a doença autoimune.
Defeitos completos foram descritos para todas as proteínas do complemento, exceto carboxipeptidase N sérica. As deficiências secundárias resultam do consumo do complemento como resultado da inflamação, autoanticorpos (por exemplo, fatores nefríticos C3/C5; e autoanticorpos contra C1q, inibidor C1 ou fator H), diminuição da síntese e/ou aumento do catabolismo ou síndromes de perda proteica. Variantes de ganho de função de determinados componentes do complemento (por exemplo, C3, C2) foram identificados e associados com a ativação excessiva do complemento em certas formas de nefropatias.[17][18]
Estados de deficiência do complemento e características associadas
Componentes
C1q: semelhante ao lúpus eritematoso sistêmico (LES) (ocorre em >90% dos pacientes com deficiência de C1q), infecções
C1r/s (principalmente combinado): semelhante a LES, artrite reumatoide (AR), infecções
C4 (C4A, C4B): semelhante ao LES, infecções (homozigoto: geralmente, clinicamente aparente; heterozigoto: normalmente, clinicamente não aparente)
C2: semelhante a LES, AR, infecções (pneumonia), vasculite; com frequência, clinicamente não evidente
C3: infecções piogênicas
C5: meningite (Neisseria), LES
C6: meningite (Neisseria), LES
C7: meningite (Neisseria), LES
C8 alfa-gama/C8 beta: meningite (Neisseria), LES
C9: infecções por Neisseria (na maioria das vezes assintomáticas)
Fator B: infecções por Neisseria
Fator D: infecções por Neisseria
Lectina ligante de manose (MBL): infecções bacterianas (na maioria das vezes, assintomáticas)
Ficolina 3 (ficolina H): infecções respiratórias, enterocolite necrosante
Serina protease 2 associada a MBL (MASP-2): infecções respiratórias.
Reguladores
Inibidor C1: angioedema hereditário
Properdina: meningite (Neisseria)
Fator H: infecções, síndrome hemolítico-urêmica atípica (SHUa)/glomerulopatia por C3 (C3G), SHUa, C3G/glomerulonefrite membranoproliferativa (GNMP)
FHR1 (FHR3): SHUa, AR, LES (com frequência associados a autoanticorpos anti-fator H e deficiência de proteínas CFHR [relacionada com o fator H do complemento], causando suscetibilidade a SHUa mediada por autoanticorpos)
Fator I: infecções (sepse, meningite, pneumonia), SHUa
CD46/MCP (proteína cofator de membrana): SHUa
CD55/DAF (fator acelerador de decaimento): enteropatia grave (enteropatia perdedora de proteínas), hemoglobinúria paroxística noturna (mutação somática do gene PIGA)
CD59: sintomas parecidos com os da síndrome de Guillain-Barré, hemólise, hemoglobinúria paroxística noturna (mutação somática do gene PIGA).
Receptores
CR3 (CD11b/CD18): deficiência de adesão leucocitária
CR4 (CD18/CD11c, LFA-1): deficiência de adesão leucocitária.
O sistema complemento compreende mais de 50 proteínas do complemento, incluindo componentes individuais do sistema em cascata, fatores reguladores do complemento (muitos dos quais são restritos à superfície das células) e receptores. Eles estão presentes no plasma e em outros fluidos corporais e também podem exercer funções imunomoduladoras intracelulares essenciais.[19]
Os genes de complemento são distribuídos em diferentes cromossomos.[20]
A maioria das proteínas do complemento é secretada pelo fígado e forma parte da resposta da fase aguda. Outros tecidos (por exemplo, rim, cérebro) também são capazes de produzir proteínas do complemento, como as células gordurosas para o fator D (adipsina). Certos componentes do complemento são proteases, que, na ativação, clivam a próxima proteína do complemento na sequência em cascata. A sequência das etapas de amplificação mostra semelhanças com a cascata de coagulação do sangue.
A cascata do complemento pode ser ativada por 3 rotas principais: a via clássica (VC), a via alternativa (VA) e a via lectina (VL).[1][Figure caption and citation for the preceding image starts]: Cascata do complemento: ativação e regulação. O complemento é ativado por meio de três vias: a via clássica, a alternativa e a de lectina. Isso resulta em: (1) geração de vários peptídeos pró-inflamatórios, que se ligam a receptores específicos em múltiplas células imunes, bem como (2) montagem do complexo de ataque à membrana, C5b-9 (CAM). Em cada nível de reação da cascata o complemento é estritamente regulado por inibidores solúveis e associados à membrana.Adaptado de uma imagem fornecida por Michael Kirschfink DVM, PhD; usado com permissão [Citation ends].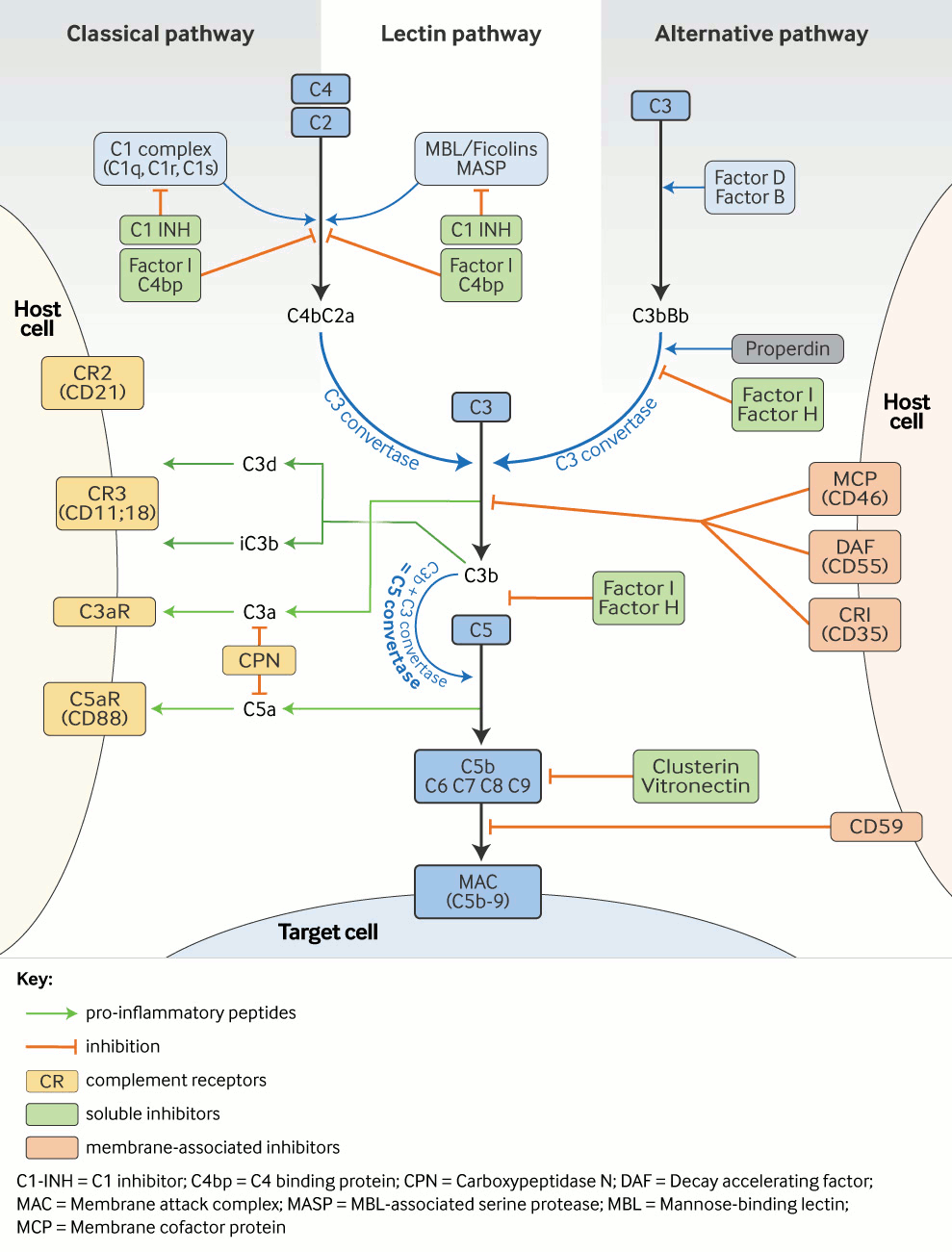
A VC complementa (melhora) respostas a anticorpos específicos, enquanto a VA e a VL, como parte do sistema imunológico inato, são importantes na defesa independente do anticorpo contra a infecção bacteriana. A terminologia do sistema dos componentes do sistema complemento se baseia nas datas de descoberta, o que explica por que a cascata não está organizada em uma ordem numérica lógica. As proteínas do complemento e os reguladores da VA se chamam fatores (por exemplo, fator B, fator H).
A ativação da VC é iniciada primariamente por mecanismos derivados de anticorpo (imunoglobulina). As regiões terminais Fc (fragmento cristalizável) dos anticorpos imunoglobulina M (IgM) e imunoglobulina G (IgG; não imunoglobulina A [IgA]) normais ativam a VC quando os anticorpos ficam estruturalmente alterados (por exemplo, quando estão vinculados a um antígeno específico dentro de um imunocomplexo ou dentro de uma crioglobulina). A VC é ativada quando C1 se liga à região Fc via sua fração C1q.
Na ausência de anticorpos, bactérias ligadas à proteína C-reativa também podem se ligar ao C1q e ativar a VC. A VL é iniciada pela ligação das ficolinas e a lectina ligante de manose (MBL, uma conhecida opsonina e um reagente de fase aguda com semelhanças estruturais com o C1q) aos resíduos de carboidrato nos patógenos e tecidos alterados. Similar ao complexo C1, a ligação da MBL ao carboidrato leva à ativação de serina proteases associadas à MBL (MSAPs) que, como os C1s, são capazes de clivar C4 e C2, conectando assim a VL à VC. Ao contrário da VC, a VA é ativada principalmente por mecanismos não anticorpo (não imunoglobulina). A hidrólise de baixo grau permanente da C3 (C3[H₂O]) leva - mediante a ligação do fator B e a subsequente clivagem pelo fator D - à geração de uma C3 convertase em fase fluida (C3b[H₂O]Bb), que é estabilizada pela properdina. Em estados saudáveis, essa atividade é autolimitada; no entanto, se a C3 recém-clivada se ligar aos patógenos ou ao tecido alterado, as respostas da VA são amplificadas. O potencial regulatório das células-alvo determina se a C3 convertase é formada na superfície, se ocorre opsonização e se a reação em cascata continua.
Assim que o complemento é ativado por qualquer uma das vias, complexos enzimáticos (C3 convertases) são gerados e clivam a C3 em 2 fragmentos (C3a e C3b). C3a é o menor fragmento. C3a é o menor fragmento e, assim como C5a, que é gerada posteriormente, é uma molécula de sinalização pró-inflamatória (anafilotoxina). Anafilatoxinas são quimioatraentes. Elas recrutam e ativam várias células inflamatórias, incluindo neutrófilos e mastócitos. Receptores para C3b e seu produto metabólico iC3b em células fagocíticas permitam a remoção dos alvos opsonizados. Imunocomplexos potencialmente patológicos (contendo anticorpo complexado com antígenos virais, bacterianos ou autoantígenos) ativam a VC e C3b, que as marca para a remoção da circulação pelos eritrócitos portadores de receptores de C3b e descarte pelas células fagocíticas no sistema reticuloendotelial. Após a ligação de C3b à C3 convertase, é gerada uma C5 convertase que cliva C5 em C5a e C5b. C5b juntamente com C6, C7, C8 e C9 geram o complexo de ataque a membrana lipofílica (CAM), C5b-9, causando a morte de célula-alvo por lise da membrana celular.
São necessárias várias proteínas regulatórias para garantir que potenciais danos teciduais mediados pelo complemento sejam evitados ou pelo menos limitados.[21] O fator H e o fator I regulam a via alternativa, enquanto o inibidor de C1 (C1-INH) e a proteína de ligação C4 (C4bp) controlam a via clássica e a via de lectina.
C3 convertases são inerentemente instáveis, com meias-vidas curtas; isso ajuda a limitar e controlar a ativação do complemento. A proteína cofator de membrana, também conhecida como MCP ou CD46, e o fator acelerador de decaimento, também conhecido como DAF ou CD55, controlam a ativação de C3 na superfície celular. A lise do complemento mediada por CAM é impedida pelos inibidores solúveis clusterina e vitronectina, e pela proteína inibidora de CAM associada à membrana (CD59).[Figure caption and citation for the preceding image starts]: Cascata do complemento: ativação e regulação. O complemento é ativado por meio de três vias: a via clássica, a alternativa e a de lectina. Isso resulta em: (1) geração de vários peptídeos pró-inflamatórios, que se ligam a receptores específicos em múltiplas células imunes, bem como (2) montagem do complexo de ataque à membrana, C5b-9 (CAM). Em cada nível de reação da cascata o complemento é estritamente regulado por inibidores solúveis e associados à membrana.Adaptado de uma imagem fornecida por Michael Kirschfink DVM, PhD; usado com permissão [Citation ends].
Existem várias deficiências do complemento hereditárias:
Via clássica: deficiência de C1q, C2, C4
Via alternativa: deficiência de fator B, fator D
Via lectina: deficiência de MBL, MASP2
Deficiência de C3
Deficiência de C5, C6, C7, C8 ou C9
Reguladores: deficiência de inibidor de C1, fator H, fator I, properdina, CD46, CD55, CD59.
deficiências do complemento representam aproximadamente 3% a 5% de todas as imunodeficiências primárias, mas podem chegar a 10%.[22] European Society for Immunodeficiencies (ESID) reporting website and interactive reporting tool Opens in new window A deficiência do complemento hereditária tem sido calculada com uma prevalência de aproximadamente 0,03%, excluindo a deficiência de lectina ligante de manose (MBL), cuja ocorrência é estimada em aproximadamente 5% da população branca geral.[22] No entanto, assim como ocorre com todos os erros inatos de imunidade/imunodeficiências primárias, um número significativo de pacientes com deficiência do complemento provavelmente permanece não diagnosticado devido à experiência clínica e laboratorial limitada das condições. As deficiências do complemento mais frequentes afetam C2 e MBL, que na maioria das vezes permanecem clinicamente silenciosas.
A incidência de angioedema hereditário (edema de Quincke) com deficiência do inibidor C1 é estimada em, aproximadamente, 1:50,000 .[23][24] No entanto, as deficiências de proteínas envolvidas na regulação do complemento são muito mais comuns em pessoas com doenças específicas. No lúpus eritematoso sistêmico, 30% dos pacientes têm uma deficiência do complemento preexistente, e acredita-se que seja de aproximadamente 20% em indivíduos com infecções por Neisseria disseminadas.[25][26][27][28][29]
A história médica pregressa pode revelar uma causa ou complicação da deficiência do complemento. A causa mais comum de deficiência do complemento adquirido é o lúpus eritematoso sistêmico (LES). No entanto, poucos pacientes com LES mostram um padrão de infecção bacteriana recorrente e, quando isso ocorre, as causas são multifatoriais e incluem uma ligação direta ao uso de agentes imunossupressores potentes no tratamento.
A forte ativação do complemento, levando frequentemente ao consumo do complemento, é vista frequentemente em pacientes com infecções graves, doenças autoimunes, vasculite, hemoglobinúria noturna paroxísmica, várias nefropatias, crioglobulinemia e lipodistrofia parcial.
As infecções manifestas devido à deficiência do complemento podem incluir:
Infecções piogênicas recorrentes (por exemplo, abscesso profundo, osteomielite, pneumonia)
Bacteremia
Infecção meningocócica recorrente
Infecção gonocócica disseminada.
As bactérias Neisseria (meningocócica e gonocócica) são particularmente sensíveis ao ataque mediado pelo complemento. Com exceção de infecções recorrentes por Neisseria, pacientes com infecções bacterianas piogênicas inexplicadas recorrentes também devem ser examinados quanto a outras deficiências imunológicas, incluindo deficiência de imunoglobulina, que é mais prevalente que a deficiência do complemento.
Alguns quadros clínicos específicos (sinais de alerta) levantam a possibilidade de deficiência do complemento.[22][30] Elas incluem:
Meningite meningocócica em indivíduos com mais de 5 anos de idade
Outras infecções bacterianas recorrentes, especialmente Pneumococcus
Angioedema sem urticária
Manifestações autoimunes
Distúrbios inflamatórios dos sistemas renal e oftálmico
Infecções gonocócicas extragenitais.
A herança normalmente é autossômica recessiva, com algumas exceções, como a deficiência de properdina (que é ligada ao X), e as deficiências de inibidor C1 e MCP/CD46 (que são autossômicas dominantes).[31] Os portadores heterozigotos geralmente se mantêm clinicamente silenciosos, exceto pela degeneração macular relacionada à idade e certas nefropatias (síndrome hemolítico-urêmica atípica, glomerulopatia do C3) Deficiências do complemento hereditárias podem ser identificadas obtendo uma história médica precisa e realizando exames laboratoriais extensivos à toda a família.
Deficiência do inibidor C1 e angioedema
O angioedema hereditário (AEH; também conhecido como edema de Quincke) representa cerca de 2% dos casos clínicos de angioedema e afeta aproximadamente 1 a cada 50,000 indivíduos.[24][32] A principal causa é a deficiência do inibidor C1.
O AEH se manifesta como edema paroxísmico agudo dos lábios, pálpebras e trato gastrointestinal (cólica) e pode ser particularmente perigoso se o edema for localizado na laringe e faringe. Obstrução intestinal, retenção urinária e cefaleia podem ser causadas por edema de outros órgãos. Estresse, infecções relacionadas a trauma e variações nos hormônios devido a gravidez, contracepção, menstruação ou terapia de reposição hormonal podem desencadear angioedema. As crises duram até 1 semana e se não forem tratadas podem levar à asfixia em 25% a 40% dos casos.[32]
Existem duas formas autossômicas dominantes de AEH por deficiência do inibidor C1. A produção inadequada do inibidor C1 (AEH-1) é reconhecida em 80% dos casos e a síntese do inibidor C1 disfuncional (AEH-2) é responsável pelos 20% restantes.[33] Ambas as formas tipicamente mostram redução de C4 como consequência secundária de atividade mal controlada da via clássica (VC). Os testes funcionais do inibidor C1 são anormais em ambos os tipos, enquanto os níveis plasmáticos de inibidor C1 são reduzidos no AEH-1, mas são normais ou elevados no AEH-2.[24][34] A bradicinina é o mediador-chave do vazamento vascular, contribuindo para o angioedema. O angioedema adquirido (AEA), muito mais raro, se desenvolve após o catabolismo excessivo do inibidor C1 ou bloqueio dos autoanticorpos.[35][36] Ele apresenta sintomas similares aos do AEH 1 e 2, e o perfil laboratorial de diagnóstico básico é indistinguível do AEH-1.[24]
Muitos pacientes apresentam manifestações clínicas similares às do AEH, mas não têm defeito do inibidor C1 e níveis normais de C4. Nesses pacientes, foram observadas mutações de ganho de função no gene FXII (FXII-AEH) e deficiências do plasminogênio, cininogênio, mioferlina e heparina sulfato-glucosamina (3-O-sulfotransferase 6).[37][38]
O inibidor C1 e C1q também pode ser consumido como parte da doença do tipo imunocomplexa maciça, como vasculite urticariforme.
Alguns casos, mas não todos, apresentam uma história familiar evidente.[39] Porém, alguns casos familiares podem não apresentar sintomas apesar de terem o mesmo defeito genético de seus parentes.
Consulte Urticária e angioedema.
Deficiência de componentes da via clássica e LES
Deficiência do complemento preexistente ou mau funcionamento dos componentes da VC, incluindo C1q, C4, C2, C3 ou receptores do complemento (incluindo o receptor de C3b), pode predispor a lúpus eritematoso sistêmico (LES). O LES mais prevalente e grave está associado com deficiência de C1, C2 ou C4.[40] Mais de três quartos dos indivíduos deficientes em uma dessas proteínas tem LES - mais de 90% nos casos de deficiência de C1q.[40] Aproximadamente 10% dos indivíduos com deficiência de C2 têm LES.[40] Os autoanticorpos contra C1q (prevalentes em uma alta porcentagem dos pacientes com LES) podem neutralizar a função do C1q e têm valor prognóstico, em particular para o desenvolvimento e a atividade de nefrite lúpica.[41] A reduzida eliminação mediada pelo complemento dos imunocomplexos e células apoptóticas é característica dessa doença e é mais pronunciada em pacientes com deficiência genética de C1q.[42]
Em determinados pacientes de LES (por exemplo, quando o LES causa glomerulonefrite, a melhora clínica frequentemente é acompanhada pela restauração dos níveis normais de C3 e C4), pode ser útil o monitoramento da função hemolítica da VC e os níveis bioquímicos de C3 e C4, dos produtos de ativação (como C4d e C3d) e do autoanticorpo anti-C1q.[41][43]
Deficiências de complemento em infecções por Neisseria disseminadas
Deficiências em C3, nos componentes terminais da via do complemento (C5-C9) e da proteína properdina da via alternativa (VA) estão fortemente associadas a um aumento na incidência de infecções meningocócicas invasivas, especialmente os sorotipos raros (W, X, Y, Z).[44] Isto é um indicativo da importância da atividade citolítica complemento na defesa do hospedeiro contra Neisseria. Para identificar essas deficiências, é obrigatório começar a análise por um rastreamento global da atividade funcional das vias clássica e alternativa usando ensaios hemolíticos (CH50, AH50) ou um ensaio de imunoadsorção enzimática (ELISA) funcional, que também inclui a via lectina. Níveis normais de AP50 e CH50 com infecção meningocócica recorrente podem indicar um defeito na properdina.
É altamente recomendável que os pacientes sejam imunizados contra Neisseria meningitidis com uma vacina conjugada tetravalente.[39][45][46][47] Deve-se observar que nem todos os sorotipos relacionados à doença são cobertos por tais vacinas.
Desregulação do complemento nas nefropatias
O complemento está implicado na patogênese da glomerulonefrite e da doença renal em estágio terminal; o rim é particularmente vulnerável à lesão mediada pelo complemento. [48][49][50] Vários tipos raros de glomerulonefrite, incluindo a doença de depósito denso (DDD) e a glomerulonefrite do C3 (C3GN), foram classificados sob o termo geral glomerulopatia do C3 (C3G).[51] Este grupo de doenças renais é causado pelo controle anormal da ativação do complemento com deposição do componente C3 do complemento nos glomérulos, levando à inflamação celular variável.[52] Os fatores da nefrite do C3 (C3Nef), que são anticorpos estabilizadores de C3, podem ser identificados em 40% a 60% dos casos de C3GN e 80% a 90% dos casos de DDD.[52] Alguns desses pacientes também desenvolvem anticorpos contra a C5 convertase (C5Nef) e, algumas vezes, também contra a VC C3 convertase (C4Nef).[53] Pacientes com C3G e, em particular os com DDD, exibem frequentemente CH50, AH50 e C3 baixos.
A síndrome hemolítico-urêmica típica (SHUa) é caracterizada por anemia hemolítica microangiopática, trombocitopenia e insuficiência renal aguda.[54] A maioria dos casos está associada com mutações ou autoanticorpos levando à ativação desregulada do complemento.[54] Ocasionalmente, mutações de genes do complemento também são encontradas na SHU diarreia-positiva, causada por Escherichia coli produtora de toxina Shiga.[54] Pacientes com SHU atípica podem ter uma história familiar positiva para SHU atípica.
O diagnóstico inclui a análise da atividade hemolítica total (CH50, AH50), C3, produtos de ativação do C3 (C3a ou C3d) e sC5b-9, bem como a análise genética molecular de C3, CFB, CFH, CHFR1-5, CFI, MCP/CD46 e THBD/CD141. Níveis reduzidos de C3 e normais de C4 indicam defeitos da via alternativa, como deficiências dos fatores H e I ou autoanticorpos (por exemplo, C3 NeF, anti-fator H) em várias formas de doenças renais (por exemplo, aHUS, DDD).
Degeneração macular relacionada à idade
A desregulação da via alternativa da cascata do complemento parece ter um papel chave na patogênese da degeneração macular relacionada à idade (DMRI). Várias proteínas do complemento, seus produtos de ativação e reguladores foram identificados nos depósitos retinianos das pessoas com DMRI. Os polimorfismos indutores de patologia nos genes que codificam as proteínas do sistema complemento, especialmente o fator H regulador, bem como C3, C2, fator B e fator I, estão associados à DMRI.[55]
Hemoglobinúria paroxística noturna
A HPN é uma doença adquirida rara, que geralmente se manifesta em adultos (mais em mulheres que em homens) com a tríade de hemólise, trombose e pancitopenia. É um distúrbio clonal dos eritrócitos causado por mutações no gene PIGA.[56] Como consequência do defeito na função do PIGA, os eritrócitos afetados não possuem proteínas de membrana ligadas ao glicosilfosfatidilinositol, incluindo CD55 e CD59. A ausência de CD59 e CD55 torna os eritrócitos da HPN susceptíveis à lise autóloga mediada pelo complemento, com a consequente anemia hemolítica. Consulte Hemoglobinúria paroxística noturna.
A deficiência de CD55 isolada é rara, mas tem sido observada em pacientes com enteropatia perdedora de proteínas grave de início precoce. Os sintomas neurológicos graves, parecidos com os da síndrome de Guillain-Barré, além da hemólise, são os sintomas característicos da deficiência de CD59 isolada.
Crioglobulinemia
A crioglobulinemia pode decorrer de neoplasia hematológica. Ela causa ativação da via clássica com complemento C4 reduzido.[57] Níveis normais de C3 e C4 não descartam crioglobulinemia. Os níveis de crioglobulina sérica devem ser testados quando a suspeita clínica de doenças associadas à redução do complemento C4 for alta (por exemplo, quando os pacientes apresentarem rash vasculítico, afetando particularmente as extremidades mais frias; artrite; e/ou insuficiência renal aguda). O teste diagnóstico definitivo envolve obter sangue coagulado fresco, coletado em um frasco térmico preaquecido (37 °C [98.6 °F]); depois o soro é rapidamente separado para reduzir o risco de a crioglobulina ficar escondida no sangue coagulado.
Lipodistrofia parcial
A lipodistrofia parcial frequentemente está associada com fator nefrítico de C3 (autoanticorpo). A estabilização da C3 convertase leva à ativação contínua do complemento e à lise dos adipócitos.[58]
Testes para uma possível deficiência do complemento devem ser realizados em pacientes com:
Doença reumática (por exemplo, lúpus eritematoso sistêmico [LES] e outras doenças do tecido conjuntivo autoimunes)
Angioedema sem urticária
Doença renal
Doença infecciosa (por exemplo, infecção bacteriana atípica ou recorrente, como casos com os sorotipos meningocócicos mais raros e casos com sepse meningocócica recorrente)
Lipodistrofia parcial
Crioglobulinemia
História familiar de deficiência do complemento.
A análise de uma possível deficiência do complemento deve começar com o rastreamento funcional de cada via de ativação, passando pela identificação do defeito nos níveis funcional, proteico e muscular.[39][59][60] Dada a importância das proteínas do complemento em uma ampla gama de sistemas biológicos, são necessários testes efetivos, padronizados e acessíveis. Existe uma grande variação na qualidade entre os laboratórios em relação ao teste do complemento e apenas alguns testes estão disponíveis comercialmente.[61] No entanto, alguns laboratórios especializados fornecem um diagnóstico abrangente, além dos parâmetros tradicionais de C3 e C4. European diagnostic complement labs Opens in new window
Amostras de sangue devem ser separadas no soro e plasma EDTA. O soro é suficiente para a análise da função total, das proteínas e reguladores do complemento e dos autoanticorpos. EDTA é absolutamente obrigatório para a quantificação dos produtos de ativação. O EDTA em uma concentração final ≥10 mM é utilizado como anticoagulante padrão, pois as propriedades complexantes do Mg²+ e Ca²+ bloqueiam a ativação in vitro do sistema complemento.[22] O sangue heparinizado e com citrato é menos útil. Se as amostras de soro/plasma não forem analisadas em algumas horas, elas devem ser congeladas e recolhidas por uma transportadoras em gelo seco, sendo enviadas para um laboratório especializado. Elas não devem ser submetidas a ciclos repetidos de congelamento-descongelamento devido ao risco de ativação in vitro.
Testes globais, como ensaios hemolíticos da via clássica (CH50) e da via alternativa (AH50), ou o ensaio da lise dos lipossomas na via clássica, fornecem informações sobre o funcionamento total da cascata do complemento. Esses testes medem artificialmente a lise da célula em alvo (eritrócito) em tubos de ensaio. Os testes dependem da formação completa dos complexos de ataque à membrana (C5 a C9) para provocar hemólise da célula em alvo. A ausência ou redução considerável da atividade indica uma deficiência do complemento primária. No entanto, esse achado também pode ser uma consequência do maior consumo do complemento levando à deficiência secundária.[22] Os níveis reduzidos de C3 e normais de C4 indicam defeitos do regulador da via alternativa (VA), como deficiências do fator H e I, ou desregulação VA induzida pelo fator nefrítico de C3, conforme observado em várias formas de nefropatias (por exemplo, síndrome hemolítico-urêmica atípica [SHUa], glomerulopatia do C3 [C3G]/doença de depósito denso [DDD]).
Para uma análise rápida da deficiência, um ensaio de imunoadsorção enzimática (ELISA) foi desenvolvido, examinando as três vias de ativação em paralelo.[62] A análise individual dos componentes é necessária subsequentemente para determinar em que parte da cascata do complemento ocorrem os defeitos. A análise dos produtos da ativação do complemento, como C3a, C3d ou sC5b-9, é necessária para distinguir as deficiências primária e secundária do complemento em virtude da hiperativação e é útil para determinar a eficácia da terapia específica do complemento (por exemplo, eculizumabe).[63]
O teste molecular se tornou essencial para esclarecer a causa genética das deficiências do complemento e está incluso, de maneira rotineira, no diagnóstico de, por exemplo, nefropatias e angioedema hereditário.[64][65]
Teoricamente, as proteínas do complemento deficiente podem ser repostas usando plasma fresco congelado (PFC) de doadores de sangue, por exemplo. Na prática isso não é geralmente recomendado, porque a meia-vida de proteínas do complemento infundido é demasiadamente curta e as proteínas podem ser pró-inflamatórias. Apesar desses problemas, o PFC foi usado com efeito positivo em alguns pacientes (por exemplo, pacientes com síndrome hemolítico-urêmica [SHU] atípica).[66] Nos casos de desregulação grave, como se pode ver na hemoglobinúria paroxística noturna e nos distúrbios renais graves, como a SHUa e a glomerulopatia do C3, os bloqueadores do complemento como o eculizumabe (anti-C5) têm sido bem-sucedidos.[67][68][69] Enquanto isso, a terapia das várias formas de angioedema foi muito além da reposição de C1-INH, e incluir ter como alvo direto o receptor de bradicinina (acetato de icatibanto) e a calicreína plasmática (ecalantide).[24][70]
As etapas do tratamento clínico genérico incluem:[39]
Rastrear parentes quanto à deficiência do complemento oculta
Incentivar e otimizar programas de imunização (buscando reforçar os níveis de anticorpos protetores quando possível). As mesmas vacinas são recomendadas para pacientes com deficiências do complemento e para indivíduos saudáveis. Algumas vacinas com foco específico (por exemplo, pneumocócica, meningocócica e Hib) são indicadas para uso fora da faixa etária recomendada, devido ao aumento do risco de doenças que podem ser evitadas pela vacina nessa população.[39][46][47][71]
Considerar a prescrição de antibióticos preventivos diariamente (por toda a vida) (semelhante à profilaxia antibiótica pós-esplenectomia).
Várias organizações e sociedades profissionais produziram diretrizes para auxiliar os profissionais da saúde no diagnóstico e manejo de deficiências do complemento e suas características associadas. Elas incluem:
European Society for Immunodeficiencies (ESID) e European Reference Network on Rare Primary Immunodeficiency, Autoinflammatory and Autoimmune Diseases (ERN RITA). Diretrizes sobre complementos da ESID e ERN RITA: deficiências, diagnóstico e manejo.[39]
Diretrizes que descrevem o sistema complemento de as funções das vias constituintes, com foco particular nas imunodeficiências primárias e seu diagnóstico e manejo.
World Allergy Organization (WAO) e European Academy of Allergy and Clinical Immunology (EAACI). Diretrizes internacionais da WAO/EAACI para o tratamento de angioedema hereditário - revisão e atualização de 2021.[24]
Diretrizes globais sobre o diagnóstico e tratamento do AEH, incluindo as principais recomendações e orientações sobre questões clínicas comuns e importantes em pacientes com AEH, com deficiência do inibidor C1 (tipo 1) e AEH com inibidor C1 disfuncional (tipo 2). As diretrizes visam a ajudar a estabelecer padrões globais para o tratamento do AEH e estimular e facilitar o uso dos diagnósticos e terapias recomendados para todos os pacientes.
Joint Task Force on Practice Parameters (JTFPP), representando a American Academy of Allergy, Asthma & Immunology (AAAAI); o American College of Allergy, Asthma & Immunology (ACAAI); e o Joint Council of Allergy, Asthma and Immunology. Atualização de parâmetros direcionada: angioedema hereditário, deficiência do inibidor C1 adquirida e angioedema associado ao inibidor da enzima conversora de angiotensina.[34]
Diretrizes dos EUA sobre o diagnóstico e tratamento do AEH.
Advisory Committee on Immunization Practices (ACIP), Centros de Controle e Prevenção de Doenças (CDC). Cronograma de vacinação de adultos: recomendações para 19 anos de idade ou mais, Estados Unidos, 2024.[47]
O cronograma de imunização de adultos de 2024 é recomendado pelo ACIP, e aprovado pelo CDC, pelo American College of Physicians, pela American Academy of Family Physicians, pelo American College of Obstetricians and Gynecologists, pelo American College of Nurse-Midwives, pela American Academy of Physician Associates e pela Society for Healthcare Epidemiology of America. Ele resume as recomendações para imunização de adultos do ACIP, além de listar as contraindicações e precauções relativas a todas as vacinas recomendadas rotineiramente no cronograma. Ele também descreve o cronograma de imunização de adultos recomendado por quadro clínico, incluindo deficiências do complemento.
Advisory Committee on Immunization Practices (ACIP), Centros de Controle e Prevenção de Doenças (CDC). Cronograma de vacinação para crianças e adolescentes: recomendações para 18 anos de idade ou menos, Estados Unidos, 2024.[46]
O cronograma de imunização de crianças e adolescentes de 2024 é recomendado pelo ACIP, e aprovado pelo CDC, pela American Academy of Pediatrics, pela American Academy of Family Physicians, pelo American College of Obstetricians and Gynecologists, pelo American College of Nurse-Midwives, pela American Academy of Physician Associates e pela National Association of Pediatric Nurse Practitioners. Ele resume as recomendações de imunização de crianças e adolescentes do ACIP, além de listar as contraindicações e precauções relativas a todos os tipos de vacinas do cronograma. Ele também descreve o cronograma de imunização de crianças e adolescentes recomendado por quadro clínico, inclusive deficiências do complemento.
Advisory Committee on Immunization Practices (ACIP), Centros de Controle e Prevenção de Doenças (CDC). Diretrizes gerais de boas práticas de imunização: imunocompetência alterada.[71]
Diretrizes que discutem as boas práticas gerais de imunização em pacientes com imunocompetência alterada, incluindo indicações de administração da vacina fora da faixa etária recomendada rotineiramente.
Grupo de trabalho para deficiência de A-1-INH. Consenso internacional e diretrizes práticas sobre o manejo ginecológico e obstétrico de pacientes do sexo feminino com angioedema hereditário causado por deficiência do inibidor C1.[72]
Diretrizes para otimização do manejo de eventos ginecológicos e obstétricos em pacientes do sexo feminino com AEH causado por deficiência de C1-INH.
International Complement Society (ICS) e International Union of Immunological Societies (IUIS). Análise do complemento de 2016: indicações clínicas, diagnósticos laboratoriais e controle de qualidade.[59]
Diretrizes sobre análise do complemento moderno e padrões para o desenvolvimento de programas de testes internacionais.
O uso deste conteúdo está sujeito ao nosso aviso legal