Detalhes
A cardiopatia congênita (CC) é a malformação congênita mais comum, embora ainda seja relativamente rara. Centers for Disease Control and Prevention: facts about congenital heart defects Opens in new window O rastreamento para CC fetal inclui ultrassonografia no segundo trimestre da gestação e exame clínico pós-parto; no entanto, as taxas de detecção são baixas.[1][2][3] A ecocardiografia fetal está se tornando o teste definitivo para o diagnóstico. A American Society of Echocardiography produziu recomendações sobre quando encaminhar para exames de imagem fetal, embora reconheça que existam diferenças locais nas indicações de encaminhamento, dependendo da população que está sendo examinada.[4]
O reconhecimento precoce de CC é importante porque o quadro clínico e a deterioração podem ser súbitos e alguns defeitos tratáveis podem causar inclusive a morte antes do diagnóstico.[1]
O tratamento cirúrgico e clínico da CC melhorou significativamente ao longo dos últimos 50 anos. As primeiras cirurgias cardíacas abertas bem-sucedidas para defeitos intracardíacos foram realizadas nos EUA na década de 1950.[5][6][7] Foi a introdução das máquinas de perfusão dos órgãos vitais, enquanto o cirurgião cuidadosamente corrigia um coração sem batimentos, que revolucionou o campo da cirurgia corretiva. Hoje espera-se uma sobrevida boa na fase adulta para a maioria dos bebês nascidos com CC.[8]
Em todo o mundo, a prevalência de cardiopatia congênita (CC) é estimada em 0.8% a 1.2% dos nascidos vivos.[9] Na América do Norte de alta renda, incluindo os EUA, a prevalência de CC no nascimento é estimada em 12.3 por 1000 (IC de 95%: 10.9 a 13.8). Estima-se que 1% ou um mínimo de 40,000 crianças por ano sejam afetadas por CC a cada ano nos EUA. Cerca de 25%, ou 2.4 por 1000 nascidos vivos, serão submetidos a tratamento invasivo no primeiro ano de vida. O tipo mais comum de defeito cardíaco congênito é a valva aórtica bicúspide, que ocorre em 13.7 em cada 1000 pessoas.[10]
Anormalidades cromossômicas (por exemplo, síndrome de DiGeorge, síndrome de Down ou síndrome de Turner) são encontradas em 8% a 10% dos indivíduos com CC e variantes no número de cópias de nucleotídeo único ou patogênicas são encontradas em 5% a 15%.[10]
Relatórios recentes mostram uma diminuição significativa na mortalidade entre pacientes com CC. Em 2020, ocorreram 2817 mortes relacionadas a CC nos EUA, uma diminuição de 11.9% em relação a 2010, e a taxa de mortalidade ajustada à idade foi de 0.9 mortes por 100,000 pessoas, uma diminuição de 18.2% em relação a 2010.[10] Devido à melhora da sobrevida, existe agora uma população idosa de adultos com CC que apresenta complicações relacionadas à doença cardiovascular adquirida (por exemplo, eventos tromboembólicos e arritmias).[8][11][12] O aumento do risco de morbidade e mortalidade por doença cardiovascular aterosclerótica (DCVA) prematura neste grupo representa um desafio crescente e destaca a necessidade de monitoramento de longo prazo dos pacientes e conscientização sobre seus fatores de risco para DCVA.[8]
Desvios circulatórios esquerda-direita
Lesões que permitem que o sangue desvie do lado esquerdo para o direito do coração. Estão associadas a níveis variados de fluxo sanguíneo pulmonar aumentado e são tipicamente acianóticas. Em alguns defeitos, o local do desvio circulatório pode não estar dentro do coração em si.
A cianose ocorre somente se as lesões forem grandes e não corrigidas na infância e se o paciente desenvolver uma doença vascular pulmonar obstrutiva (fisiologia de Eisenmenger). A ecocardiografia é a principal modalidade de diagnóstico por imagem e, atualmente, o papel do cateterismo cardíaco é essencialmente intervencionista.[13]
Exemplos incluem:
Defeito do septo ventricular (DSV)
Defeito do septo atrial (DSA)
Defeito do septo atrioventricular (DSAV)
Persistência do canal arterial (PCA)
Conexão venosa pulmonar anômala parcial (CVPAP).
Desvios circulatórios direita-esquerda
Lesões que resultam em sangue não oxigenado alcançando a aorta e estão associadas ao aumento ou à diminuição do fluxo sanguíneo pulmonar.
Exemplos incluem:
Tetralogia de Fallot
Atresia da valva pulmonar com ou sem um DSV
Dextrotransposição das grandes artérias (DTGA)
Tronco arterioso
Anomalia de Ebstein
Conexão venosa pulmonar anômala total (CVPAT)
Síndrome do coração esquerdo hipoplásico (SCEH).
Lesões valvares e não valvares obstrutivas
Obstrução da via de saída do ventrículo esquerdo (VSVE)
Coarctação da aorta
Estenose pulmonar valvar (EPV)
Estenose da valva aórtica (EA)
Os defeitos cardíacos congênitos também podem ser classificados usando a classificação anatômica/fisiológica (AP) introduzida pela força-tarefa da American Heart Association/American College of Cardiology (AHA/ACC) em suas diretrizes de 2018.[14]
No sistema AP, um paciente é classificado tanto com uma classe "A" quanto com uma classe "P" com base na característica anatômica ou fisiológica relevante "mais importante", e os pacientes podem passar de uma classificação AP para outra ao longo do tempo. Se o quadro clínico se agravar, a classificação mudará para um grupo de maior gravidade, mas a melhora do quadro; por exemplo, após uma intervenção como substituição de valva ou controle de arritmia, pode resultar na mudança para uma classificação de gravidade mais baixa.
Nas diretrizes de 2020 para o manejo de adultos com cardiopatias congênitas publicadas pela European Society of Cardiology (ESC), apenas a classificação anatômica é utilizada, sem o componente fisiológico.[15] Existem diferenças na classificação de gravidade em ambos os sistemas de classificação; por exemplo, a estenose congênita da valva aórtica e a valvopatia mitral congênita são consideradas defeitos leves nas diretrizes da ESC, enquanto as diretrizes da AHA/ACC os classificam como moderados.
O DSV é a forma mais comum de CC, responsável por 20% de todos os casos, com exceção do prolapso da valva aórtica bicúspide e da valva mitral.[16] Os subtipos baseados na localização incluem: perimembranoso (na região da membrana septal); saída (abaixo de 1 ou ambas as valvas semilunares); entrada (inferior às valvas atrioventriculares [AV]); e muscular (na porção muscular ou trabeculada do septo ventricular).[17][Figure caption and citation for the preceding image starts]: Subtipos dos defeitos do septo ventricular: (A) saída; (B) perimembranoso; (c) entrada; (D) muscularMayo Clinic Foundation [Citation ends].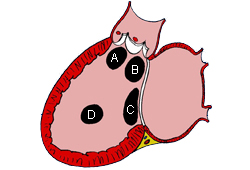
Os defeitos perimembranosos normalmente ocorrem como uma lesão solitária e, às vezes, podem fechar espontaneamente pela aposição do folheto septal da valva tricúspide ao defeito. Os defeitos de saída podem ser grandes e associados a formas mais complexas de cardiopatias congênitas, como a tetralogia de Fallot. Os DSVs de saída ou peri-membranosos estão intimamente próximos da cúspide direita da valva aórtica. Por causa do efeito Venturi, esses defeitos podem causar prolapso de uma cúspide da valva aórtica, o que resulta em uma restrição do fluxo através do DSV e regurgitação da valva aórtica.[18] Os defeitos de entrada não se fecham espontaneamente e podem estar associados com o defeito do septo atrioventricular (DSAV) e com a regurgitação da valva AV. Os defeitos musculares são os tipos mais comuns de DSVs em neonatos e a grande maioria fecha espontaneamente antes dos 2 anos de idade.[Figure caption and citation for the preceding image starts]: Imagem da ecografia apical de 4 câmaras de um defeito do septo ventricular (DSV) muscular (seta). (AD) átrio direito; (AE) átrio esquerdo; (VD) ventrículo direito; (VE) ventrículo esquerdoImagem cedida por Patrick W. O'Leary, MD [Citation ends].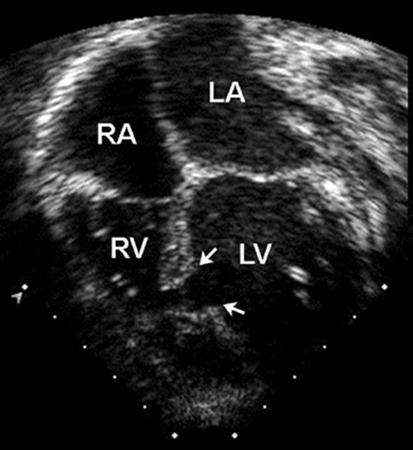
Crianças com DSV pequeno se desenvolvem normalmente e são assintomáticas. Com defeitos maiores, o excesso de fluxo sanguíneo pulmonar que ocorre após as 6-8 semanas de idade, quando ocorre a diminuição normal do fluxo sanguíneo pulmonar, pode levar ao desenvolvimento de taquipneia, taquicardia, palidez, baixa aceitação alimentar e baixo ganho de peso. A remodelação vascular pulmonar e a doença obstrutiva geralmente não se desenvolvem em bebês, mas podem se desenvolver até os 2 anos de idade. Se não forem tratados, a maioria dos DSVs moderados a grandes acabam causando hipertensão pulmonar grave e, em algum momento, ocorre a reversão do shunt, levando à cianose central. Essa condição, chamada síndrome de Eisenmenger, normalmente não ocorre até mais tarde na vida. Uma vez que o paciente desenvolva a fisiologia de Eisenmenger, o fechamento do DSV é contraindicado.[19][20]
Os pacientes apresentam um sopro holossistólico de frequência baixa a média, um impulso pré-cordial proeminente e, em pacientes com hipertensão pulmonar, uma segunda bulha cardíaca alta e única. Se o desvio circulatório esquerda-direita for grande, os pacientes desenvolvem sopro ("ruflar") do fluxo mesodiastólico através da valva mitral.
A radiografia torácica e o eletrocardiograma (ECG) podem estar normais nos DSVs. Nos defeitos maiores, há cardiomegalia e aumento das marcas vasculares pulmonares na radiografia torácica, e o ECG revela hipertrofia do ventrículo esquerdo; a hipertrofia do ventrículo direito pode ocorrer em defeitos maiores. Os DSVs de entrada estão associados ao desvio do eixo esquerdo no ECG. A ecocardiografia fornece informações importantes sobre a anatomia do defeito, o volume do desvio circulatório e a pressão do ventrículo direito.[21][22][Figure caption and citation for the preceding image starts]: Radiografia torácica demonstrando a transposição circulatória pulmonarMayo Clinic Foundation [Citation ends].
O objetivo do tratamento é assegurar um crescimento somático adequado e evitar a doença vascular pulmonar obstrutiva. Na ausência de desenvolvimento de hipertensão pulmonar, os DSVs normalmente causam o aumento do ventrículo esquerdo (VE), o que serve como indicação para fechamento. Os DSVs pequenos com fluxo restritivo podem não ser tratados, sendo necessária vigilância contínua para o desenvolvimento de aumento do VE e endocardite. Os DSVs grandes e os defeitos associados à regurgitação aórtica decorrente do prolapso da cúspide da valva aórtica ou do excesso de fluxo sanguíneo pulmonar podem precisar de diuréticos e uma fórmula calórica aumentada, seguidos por fechamento do defeito (por fechamento cirúrgico ou transcateter). Aqueles com aumento do fluxo sanguíneo pulmonar e deficit no crescimento são fechados cirurgicamente entre 1 e 4 meses de idade; a maioria dos defeitos grandes é tipicamente fechada entre os 6 e 9 meses de idade. Para pacientes com síndrome de Eisenmenger, a terapia vasodilatadora pulmonar é o tratamento de primeira escolha, e as mulheres em idade fértil devem ser informadas de que a gravidez não é recomendada devido à alta mortalidade materna e às complicações fetais.[19][20]
Para obter mais informações, consulte Defeitos do septo ventricular.
As comunicações interatriais incluem defeitos do septo atrial (DSA) e outros shunts pré-tricúspide (por exemplo, defeito do seio venoso e seio coronário sem teto). Os shunts pré-tricúspide são definidos como a presença de um shunt esquerda-direita que ocorre proximal à valva atrioventricular direita. Em comparação com shunts pós-tricúspide (como defeito do septo ventricular e persistência do canal arterial), esses defeitos são caracterizados por shunt de baixa pressão e grau variável do volume do shunt. Os shunts pré-tricúspide representam 6% a 10% de todas as CC e têm uma predominância de mulher para homem de 2:1.[23] Os principais tipos consistem do defeito do septo atrial ostium secundum (60% a 70% de todos os shunts pré-tricúspide), que ocorre na área da fossa oval; defeito ostium primum, uma forma de defeito do septo atrioventricular (DSAV), no aspecto inferior do septo atrial; defeitos do seio venoso superior e inferior (nos quais o septo interatrial está intacto), que estão presentes no local de entrada da veia cava superior e inferior, respectivamente, para o átrio direito; seio coronário sem teto, o tipo menos comum.[Figure caption and citation for the preceding image starts]: Subtipos de defeitos do septo atrial. (A) seio venoso; (B) ostium secundum; (C) ostium primum; (D) seio coronário sem tetoMayo Clinic Foundation [Citation ends].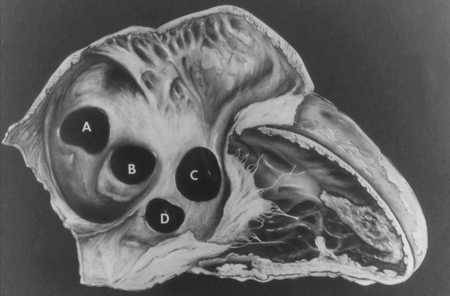 [Figure caption and citation for the preceding image starts]: Imagem da ecografia apical de 4 câmaras de um defeito do septo atrial (DSA) do ostium primum (seta). (AD) átrio direito; (AE) átrio esquerdo; (VE) ventrículo esquerdoImagem cedida por Patrick W. O'Leary, MD [Citation ends].
[Figure caption and citation for the preceding image starts]: Imagem da ecografia apical de 4 câmaras de um defeito do septo atrial (DSA) do ostium primum (seta). (AD) átrio direito; (AE) átrio esquerdo; (VE) ventrículo esquerdoImagem cedida por Patrick W. O'Leary, MD [Citation ends].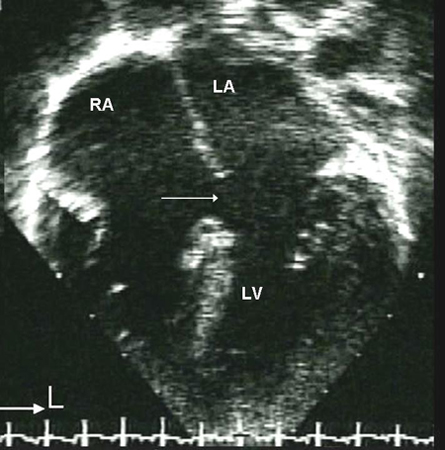
O seio coronário sem teto não é um defeito do septo atrial verdadeiro. Ocorre quando o processo de cobertura do seio coronário na vida fetal está incompleto, permitindo a comunicação entre o seio coronário e o átrio esquerdo.
Crianças com shunts pré-tricúspide isolados são frequentemente assintomáticas.[24] É o diagnóstico errado de CC mais comum na infância, frequentemente não descoberto até a fase adulta. Defeitos não tratados de tamanho moderado a grande levam à intolerância ao exercício, arritmias atriais e aumento do fluxo sanguíneo pulmonar. Um aumento real na resistência vascular pulmonar é muito mais raro do que nos shunts pós-tricúspide e ocorre mais tarde na vida, mais comumente em subpopulações específicas, como aquelas com síndrome de Down. A embolização paradoxal através do defeito ocorre quase exclusivamente em pacientes com DSA secundum.
Em pacientes com defeitos moderados a grandes, o exame revela aumento do impulso ventricular direito, segunda bulha cardíaca fixa amplamente dividida e sopro sistólico de ejeção suave, melhor audível na borda esternal superior esquerda, causado pelo aumento do fluxo sanguíneo através da valva pulmonar, em vez de através do DSA. Em grandes defeitos, um sopro diastólico também pode estar presente na borda esternal inferior secundário ao fluxo sanguíneo excessivo através da valva tricúspide.[24]
A radiografia torácica não é útil na determinação do subtipo e pode ser normal em um DSA pequeno. O eletrocardiograma (ECG) também pode estar normal no secundum pequeno, seio venoso e DSAs do seio coronário sem teto. Com um desvio circulatório maior, pode haver aumento do átrio direito, hipertrofia ventricular direita ou desvio do eixo direito.[Figure caption and citation for the preceding image starts]: Imagem de ecocardiografia do eixo paraesternal curto revelando um aumento ventricular direito em um paciente com defeito do septo atrial (DSA). (VD) ventrículo direito; (VE) ventrículo esquerdoImagem cedida por Patrick W. O'Leary, MD [Citation ends].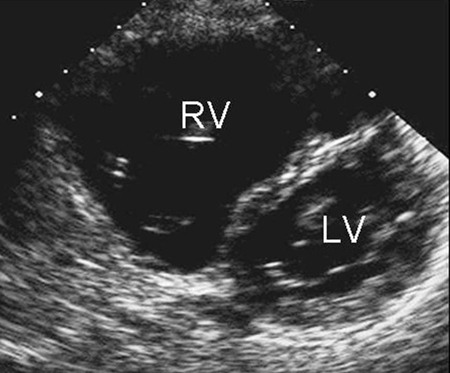 [Figure caption and citation for the preceding image starts]: Imagem de ecocardiografia apical de 4 câmaras revelando dilatação do ventrículo direito em um paciente com defeito do septo atrial (DSA). (AD) átrio direito; (VD) ventrículo direito; (VE) ventrículo esquerdoImagem cedida por Patrick W. O'Leary, MD [Citation ends].
[Figure caption and citation for the preceding image starts]: Imagem de ecocardiografia apical de 4 câmaras revelando dilatação do ventrículo direito em um paciente com defeito do septo atrial (DSA). (AD) átrio direito; (VD) ventrículo direito; (VE) ventrículo esquerdoImagem cedida por Patrick W. O'Leary, MD [Citation ends].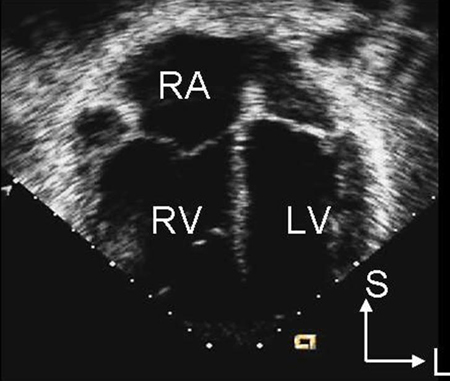 Defeitos do tipo ostium primum são caracterizados por uma rotação inicial do plano frontal no sentido anti-horário e desvio do eixo esquerdo.[24]
Defeitos do tipo ostium primum são caracterizados por uma rotação inicial do plano frontal no sentido anti-horário e desvio do eixo esquerdo.[24]
O tratamento envolve cirurgia ou fechamento por meio de um dispositivo percutâneo.[25][Figure caption and citation for the preceding image starts]: Imagem de ecocardiografia transesofágica de um dispositivo de oclusão de defeito do septo atrial (DSA) (seta). (AD) átrio direito; (AE) átrio esquerdo; (VCS) veia cava superiorImagem cedida por Patrick W. O'Leary, MD [Citation ends].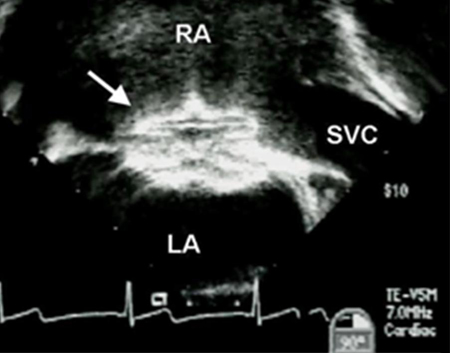 O fechamento por dispositivo é o método preferencial para defeitos do tipo ostium secundum se o defeito for de tamanho adequado (o maior dispositivo de fechamento disponível tem 40 mm de diâmetro) e se houver bordas septais adequadas para fixar o dispositivo.[26] Na maioria dos outros subtipos, o fechamento por dispositivo não é viável devido à falta de bordas e à proximidade de outras estruturas cardíacas, embora relatórios recentes sugiram o fechamento por dispositivo bem-sucedido de defeitos do seio venoso superior anatomicamente ajustados.[27] O DSAV parcial (DSA ostium primum e uma fenda da valva mitral) pode ser corrigido após os 18-24 meses de idade na maioria dos pacientes e envolve o fechamento da fenda mitral, além do DSA.
O fechamento por dispositivo é o método preferencial para defeitos do tipo ostium secundum se o defeito for de tamanho adequado (o maior dispositivo de fechamento disponível tem 40 mm de diâmetro) e se houver bordas septais adequadas para fixar o dispositivo.[26] Na maioria dos outros subtipos, o fechamento por dispositivo não é viável devido à falta de bordas e à proximidade de outras estruturas cardíacas, embora relatórios recentes sugiram o fechamento por dispositivo bem-sucedido de defeitos do seio venoso superior anatomicamente ajustados.[27] O DSAV parcial (DSA ostium primum e uma fenda da valva mitral) pode ser corrigido após os 18-24 meses de idade na maioria dos pacientes e envolve o fechamento da fenda mitral, além do DSA.
Para obter mais informações, consulte Comunicações interatriais.
Representa 4% a 5% de todas as CCs e é encontrado em 40% das crianças com síndrome de Down.[28] Esse defeito também é chamado de defeito do coxim endocárdico ou defeito do canal atrioventricular (AV). Os coxins endocárdicos fecham o ostium primum e formam porções das valvas AVs e do septo ventricular. Os DSAVs podem ser parciais ou completos; um tipo de DSAV parcial é um defeito do septo atrial (DSA) primum. Um DSAV completo consiste em um DSA primum e um defeito do septo ventricular (DSV) de entrada contígua.[Figure caption and citation for the preceding image starts]: Imagem da ecografia apical de 4 câmaras de um defeito do septo atrioventricular (DSAV) completo. Note o ostium primum no defeito do septo atrial (DSA) (*) e o defeito do septo ventricular (DSV) de entrada adjacente (seta). (AD) átrio direito; (AE) átrio esquerdo; (VD) ventrículo direito; (VE) ventrículo esquerdoImagem cedida por Patrick W. O'Leary, MD [Citation ends].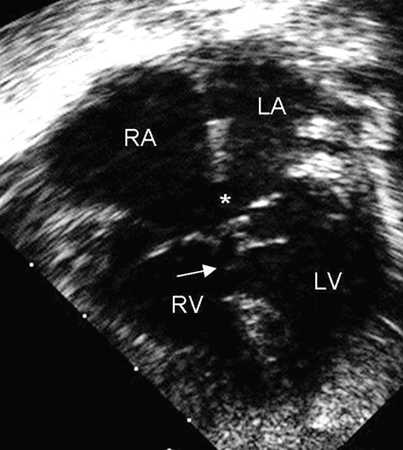
A apresentação e os achados do exame físico de pacientes com DSAVs completos e parciais são semelhantes aos dos pacientes com um DSV ou DSA, respectivamente. A gravidade também é determinada pelo volume de desvio circulatório, extensão das anormalidades da valva AV, anormalidades cardíacas e extracardíacas associadas e tamanho relativo dos 2 ventrículos.
O eletrocardiograma (ECG) provavelmente revelará uma rotação inicial anti-horária do plano frontal e um eixo QRS superior.[Figure caption and citation for the preceding image starts]: Eletrocardiograma (ECG) de 12 derivações em um lactente com defeito do septo atrioventricular (DSAV) completo; o ECG é significativo para desvio do eixo esquerdoMayo Clinic Foundation [Citation ends]. Os pacientes com DSAVs geralmente apresentam cardiomegalia e aumento das marcas vasculares pulmonares na radiografia torácica.[29]
Os pacientes com DSAVs geralmente apresentam cardiomegalia e aumento das marcas vasculares pulmonares na radiografia torácica.[29]
As crianças com um DSAV completo requerem uma correção cirúrgica, geralmente realizada nos 3-6 meses de idade. Elas precisam de acompanhamento pela vida toda, uma vez que 15% dos pacientes desenvolvem uma regurgitação progressiva da valva AV ou obstrução da via de saída do ventrículo esquerdo.[14]
O canal arterial geralmente fecha alguns dias após o nascimento e é um componente essencial da fisiologia cardiovascular intrauterina normal. A patência persistente ocorre em 1 a cada 2000 lactentes nascidos vivos a termo, mas é muito mais comum nos neonatos prematuros.[30] Nos lactentes prematuros, a PCA tem sido observada durante a internação no hospital em 42% dos neonatos pesando 500 a 999 g, 21% pesando 1000 a 1499 g e 7% pesando 1500 a 1750 g.[31] Há também uma incidência 30 vezes maior em pacientes nascidos em altitudes mais elevadas. A PCA representa 9% a 12% de todas as cardiopatias congênitas (CCs).
Os pacientes são assintomáticos quando o canal é pequeno. Conforme crescem, os neonatos apresentam sinais de fluxo sanguíneo pulmonar aumentado e pressão arterial e pulsos amplos. A fisiologia de Eisenmenger, secundária à resistência vascular pulmonar elevada e à reversão do shunt, pode ocorrer se a PCA for grande e de longa duração, e inicialmente resulta em cianose apenas na metade inferior do corpo, com aumento gradual da mistura sistêmica e cianose à medida que a doença evolui.[20]
Tipicamente, a PCA produz um sopro contínuo, mais bem auscultado na região infraclavicular esquerda. O sopro é contínuo porque a pressão aórtica é maior que a pressão arterial pulmonar durante as sístoles e diástoles. Se o desvio circulatório for grande, o sopro do fluxo mitral diastólico também pode ser auscultado. Em alguns lactentes prematuros e neonatos, o sopro pode ser auscultado apenas na sístole e pode ser confundido com o sopro de um defeito do septo ventricular (DSV).
Com frequência, o ECG e a radiografia torácica são normais. Em uma PCA grande, podem estar presentes hipertrofia biventricular na superfície do ECG e aumento do fluxo sanguíneo pulmonar e cardiomegalia na radiografia torácica. A ecocardiografia permite delinear a anatomia da PCA e a direção e volume do desvio circulatório.[32] Uma avaliação mais aprofundada da anatomia e da proximidade com outras estruturas pode ser obtida com exames transversais, como um estudo de tomografia computadorizada.[22]
Em lactente prematuros, o canal pode frequentemente ser fechado com indometacina ou ibuprofeno ou, se isso falhar, ligado cirurgicamente.[33] Em crianças mais velhas e adultos, na ausência de hipertensão arterial pulmonar significativa, as PCAs são fechadas por via percutânea por meio de espirais ou dispositivos.[14][26] Diuréticos pode ser usados para tratar pacientes com excesso de fluxo sanguíneo pulmonar até o fechamento definitivo.
Para obter mais informações, consultePersistência do canal arterial.
Uma anomalia na qual ≥1 (mas não todas) veias pulmonares se conectam às veias sistêmicas ou átrio direito, em vez de ao átrio esquerdo. Esse defeito representa <1% de todas as CCs. A conexão anômala da(s) veia(s) pulmonar(es) superior/média direita(s) está associada a um defeito do seio venoso; a conexão da(s) veia(s) pulmonar(es) inferior(is) direita(s) à veia cava inferior pode fazer parte de um defeito do seio venoso inferior, também conhecido como “síndrome de cimitarra”.[34]
Os achados na apresentação, no ECG e na radiografia torácica são semelhantes aos dos paciente com DSAs, embora os pacientes com apenas uma veia anormal possam ser, com frequência, clinicamente assintomáticos. O quadro clínico dos pacientes com síndrome de cimitarra varia de adultos relativamente assintomáticos a crianças gravemente enfermas com hipoplasia pulmonar ipsilateral, infecções do trato respiratório e hipertensão pulmonar.[35]
A ecocardiografia transtorácica pode não ser suficiente para diagnosticar a conexão venosa pulmonar anômala parcial (CVPAP) de maneira acurada em adultos, e outros modalidades de diagnóstico por imagem como a ecocardiografia transesofágica, tomografia computadorizada (TC) ou ressonância nuclear magnética (RNM) podem ser necessárias.
A intervenção cirúrgica será indicada se o volume do desvio circulatório criar uma sobrecarga de volume significativa do lado direito, a qual está associada ao sopro do fluxo de saída pulmonar e, por vezes, ao sopro do fluxo de entrada diastólico da tricúspide.[24]
A tetralogia de Fallot é a CC cianótica mais comum, representando 4% a 8% de todos os defeitos, e pode ocorrer como uma lesão cardíaca solitária ou como parte de uma síndrome genética, a mais comum das quais é a síndrome de DiGeorge (uma microdeleção de 22q11.2 ).[36] Ela consiste em 4 anormalidades: defeito do septo ventricular (DSV) de saída, obstrução da via de saída do ventrículo direito (VSVD), uma aorta que se sobrepõe à crista septal ventricular e hipertrofia do VD, um resultado da obstrução da VSVD.[Figure caption and citation for the preceding image starts]: Imagem de ecocardiografia do eixo paraesternal longo em um paciente com tetralogia de Fallot. A aorta (Ao) sobrepõe-se ao defeito do septo ventricular (DSV) (*). (AE) átrio esquerdo; (VD) ventrículo direito; (VE) ventrículo esquerdoImagem cedida por Patrick W. O'Leary, MD [Citation ends].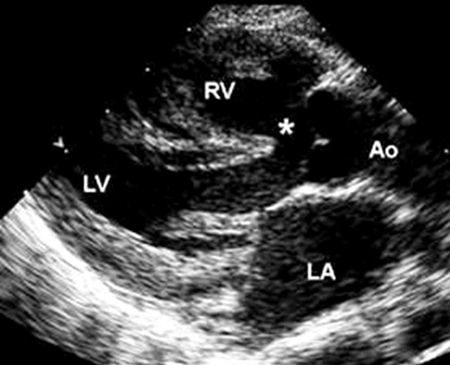
Se a obstrução da VSVD for leve ou não houver shunt direita-esquerda e cianose aparente, esse quadro fisiológico é, às vezes, chamado de "tetralogia rosada". A maioria dos pacientes com tetralogia de Fallot, contudo, apresenta cianose progressiva após o nascimento, seguida por dispneia ao esforço na primeira infância. Uma eritrocitose secundária se desenvolve como um mecanismo compensatório para a hipoxemia crônica. Um crescimento somático deficiente pode estar presente. Crises hipercianóticas são episódios hipóxicos que consistem de um início abrupto de respiração superficial rápida, agitação aumentada, cianose e uma diminuição da intensidade do sopro devido ao fluxo de sangue reduzido através da VSVD. Sem tratamento, estes podem causar hipoxemia profunda, convulsões e morte. Crianças mais velhas com crises de hipóxia agacham-se para aumentar a resistência vascular sistêmica e, assim, reduzir a hipoxemia.
Um sopro sistólico de ejeção alto é auscultado na borda esternal superior esquerda, irradiando para ambas as axilas devido à obstrução da VSVD. Raramente, os pacientes com obstrução mínima da VSVD apresentam apenas uma cianose leve e podem desenvolver sinais de fluxo sanguíneo pulmonar excessivo devido ao desvio circulatório esquerda-direita através do DSV. Um impulso ventricular direito pode estar presente.
Classicamente, a radiografia torácica é normal, exceto pelas marcas vasculares pulmonares diminuídas. Menos de 25% dos lactentes apresentam a classicamente descrita silhueta cardíaca em "forma de bota".
Os lactentes gravemente hipóxicos podem ser tratados com prostaglandina E1 para reabrir ou manter a permeabilidade do canal arterial e aumentar o fluxo sanguíneo pulmonar. Se isso não melhorar a hipoxemia, o paciente pode precisar de um enxerto arterial sistêmico-pulmonar, como uma anastomose Blalock-Taussig modificada, até a correção definitiva (que inclui fechamento com enxerto do DSV e alívio da obstrução da VSVD).[37] A correção completa pode ser realizada em determinados centros no período neonatal, embora a maioria dos médicos prefira fazê-lo aos 3 meses de idade ou quando a cianose progressiva se desenvolver. Os resultados cirúrgicos têm sido excelentes nas últimas décadas, embora muitos pacientes reparados quando crianças necessitam de reimplante de uma valva pulmonar competente quando mais velhos, o que pode ser realizado cirurgicamente ou através de um procedimento transcateter.[14]
Exames laboratoriais de acompanhamento são essenciais para identificar a necessidade oportuna de substituição da valva, o tipo de tratamento necessário e para monitorar o desfecho do tratamento.[38] As arritmias ventriculares são uma complicação potencial de risco de vida em longo prazo da tetralogia de Fallot reparada, com a etiologia provável sendo istmos anatômicos de condução lenta criados no momento do reparo cirúrgico original. O tratamento inclui ablação com ou sem cardioversor desfibrilador intracardíaco.[39]
Para obter mais informações, consulte Tetralogia de Fallot.
Esta é frequentemente considerada a forma mais grave de tetralogia de Fallot, na qual a valva pulmonar não se desenvolve. Setenta por cento dos pacientes apresentam persistência do canal arterial (PCA) que mantém o fluxo sanguíneo pulmonar. Outros apresentam vários vasos sistêmico-pulmonares colaterais. As anormalidades arteriais pulmonares são comuns e incluem hipoplasia, não confluência e distribuição anormal.[40]
Os pacientes são geralmente cianóticos ao nascimento. Um sopro contínuo da PCA ou colateral das artérias sistêmico-pulmonares pode ser audível. A segunda bulha cardíaca é única. O ECG revela desvio do eixo direito e hipertrofia ventricular direita. A radiografia torácica revela uma diminuição significativa da vascularidade pulmonar.
A prostaglandina E1 entra em ação para manter a permeabilidade do canal arterial. A maioria dos pacientes também provavelmente precisará de um desvio circulatório sistêmico-pulmonar para manter um fluxo sanguíneo pulmonar adequado. Os pacientes com artérias pulmonares confluentes e grandes podem necessitar de uma correção em estágio único; outros precisam de uma correção em estágios. Pacientes com múltiplos colaterais das artérias sistêmico-pulmonares correm risco de desenvolver hipertensão pulmonar precoce, que inicialmente pode ser segmentar.[20]
Esse defeito é raro e a gravidade depende das hipoplasias do ventrículo direito e da valva tricúspide comumente associadas. Fístulas da artéria coronária-ventrículo direito também estão associadas com esse defeito.[41] Um defeito do septo atrial (DSA) grande e não restritivo é necessário para sobrevivência, uma vez que não há saída do ventrículo direito para as artérias pulmonares.
Não há achados no exame físico característicos desse defeito. A radiografia torácica revela cardiomegalia com um átrio direito proeminente e o eletrocardiograma (ECG) revela um aumento atrial. A ecocardiografia é usada para estabelecer o tamanho da hipoplasia do ventrículo direito e da valva tricúspide.
Um DSA restritivo é uma emergência médica nesses pacientes e requer uma septostomia atrial por balão de urgência. Os pacientes devem ser tratados com prostaglandina E1 para manter a permeabilidade do canal, uma vez que a persistência do canal arterial é a única fonte de fluxo sanguíneo pulmonar.[42][43]
A maioria dos pacientes precisa de cirurgias em estágios que potencialmente permitem que a valva tricúspide e ventrículo direito aumentem em tamanho. Uma operação de Fontan modificada ou uma correção ventricular 1.5 podem ser necessárias se o ventrículo direito e a valva tricúspide não alcançarem um tamanho adequado para permitir uma correção ventricular 2. Uma correção ventricular 1.5 envolve um desvio circulatório cavo-pulmonar bidirecional (anastomose de "Glenn") para desviar o fluxo sanguíneo da veia cava superior para as artérias pulmonares para reduzir o pré-preenchimento do ventrículo direito, fechamento do DSA e estabelecimento da continuidade do ventrículo direito-artéria pulmonar. Os pacientes com fístulas da artéria coronária-ventrículo direito podem requerer uma operação de Fontan modificada.
Uma CC comum, representando 4% de todos os defeitos. Nesse defeito, a aorta surge do ventrículo direito e a artéria pulmonar do ventrículo esquerdo. Como resultado, sangue venoso não oxigenado sistêmico percorre o ventrículo direito até a aorta sem passar através dos pulmões, e as veias pulmonares percorrem o ventrículo esquerdo até os pulmões por meio da artéria pulmonar. As circulações sistêmicas e pulmonares estão em paralelo, e não em série. Isso não é compatível com a vida sem comunicação entre os 2 circuitos, como defeito do septo ventricular (DSV), defeito do septo atrial (DSA) ou persistência do canal arterial (PCA). A maioria dos pacientes com DTGA não apresenta um DSV, e um terço daqueles com DSV apresenta uma estenose pulmonar associada.
Os pacientes se apresentam nas primeiras horas após o nascimento com cianose como uma emergência médica.
O exame físico revela um impulso ventricular direito proeminente, uma segunda bulha cardíaca única e alta (decorrente da posição anterior da aorta) e nenhum sopro ou um sopro sistólico de ejeção nos pacientes com um DSV e estenose pulmonar.
A prostaglandina E1 é usada para manter a permeabilidade do canal arterial, e a maioria dos pacientes também requer uma septostomia atrial para melhorar a mistura no nível atrial. A operação de troca arterial, incluindo reimplante de botão de artéria coronária, é a melhor opção para pacientes com DTGA e valva pulmonar normal, e é realizada nas primeiras semanas de vida. Uma vez que a resistência vascular pulmonar diminui, o ventrículo esquerdo não é mais condicionado a bombear contra a resistência maior associada à circulação sistêmica. O TGA-DSV deve ser corrigido no período neonatal, se identificado na época. Contudo, foi relatado uma correção tardia (1-2 meses de idade) bem-sucedida, embora com morbidade aumentada. Pacientes com a combinação de TGA, DSV e estenose pulmonar podem ter sido submetidos a uma operação de Rastelli. Esta operação usa um enxerto de DSV angulado para direcionar o sangue do ventrículo esquerdo (VE) através do DSV para a aorta anterior. Este procedimento também envolve a colocação de um conduto do ventrículo direito (VD) até a artéria pulmonar (AP). Embora essa operação resulte na restauração do VE como ventrículo sistêmico, o manejo em longo prazo geralmente envolve múltiplas reoperações para substituição do conduto VD-AP. Embora não seja mais utilizado, principalmente adultos mais velhos (>35-40 anos de idade) nascidos com DTGA foram submetidos ao procedimento de troca atrial, seja na forma de operação de Senning ou de Mustard.[38] O VD é o ventrículo sistêmico após esses procedimentos.[26] Cerca de 80% desses pacientes tiveram bons desfechos várias décadas após a cirurgia. No entanto, complicações em longo prazo, incluindo arritmias e anomalias de condução, obstrução ou regurgitação venosa sistêmica ou pulmonar e disfunção ventricular direita sistêmica resultando em insuficiência cardíaca, ocorrem na grande maioria dos pacientes adultos.[14] Adultos que apresentam disfunção do VD após procedimentos de troca atrial são candidatos ao transplante cardíaco.[26]
Envolve <1% de todas as CCs. Consiste de um grande defeito do septo ventricular (DSV) de saída e apenas 1 grande artéria, o tronco, surgindo do coração, de onde originam-se as artérias coronárias, pulmonares e a aorta. Existem quatro subtipos, baseados na presença ou ausência de uma artéria pulmonar principal, na ramificação das artérias pulmonares e na formação da aorta.[44]
[Figure caption and citation for the preceding image starts]: Subtipos de tronco arterioso com defeito do septo ventricular (tipo A): Tipo A1: a artéria pulmonar principal está presente e se bifurca nas artérias pulmonares esquerda e direita. Tipo A2: as artérias pulmonares dos ramos direito e esquerdo surgem de um tronco comum. Tipo A3: um ramo da artéria pulmonar surge do tronco comum e o outro está ausente/surge de uma PCA ou da aorta. Tipo A4: tronco com hipoplasia do arco aórtico, coarctação ou arco aórtico interrompido e grande PCACalder L et al. Am Heart J 1976 Jul;92(1):23-38; usado com permissão [Citation ends].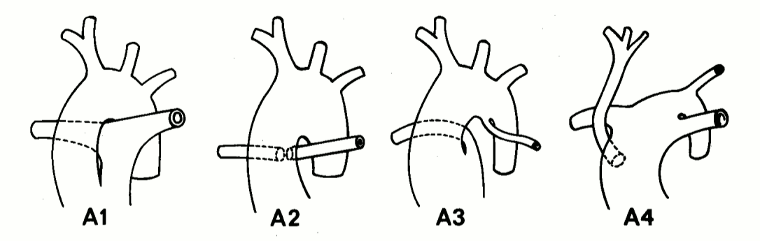
Uma microdeleção do 22q11.2 (síndrome de DiGeorge), arco aórtico interrompido e insuficiência da valva troncular são anomalias comuns.
Os pacientes apresentam insuficiência cardíaca congestiva (ICC) ou cianose.
O exame físico revela um impulso ventricular direito proeminente e um sopro sistólico de ejeção na borda esternal esquerda. Pode haver um clique de ejeção da valva troncular. A regurgitação da valva troncular resulta em um sopro diastólico em decrescendo. A interrupção da aorta e uma persistência do canal arterial (PCA) restritiva resultam em diminuição dos pulsos femorais.
A dominância ventricular direita é observada na primeira infância no ECG, e a radiografia torácica revela cardiomegalia com aumento das marcas pulmonares. A ecocardiografia transtorácica auxilia na identificação mais precisa de vários aspectos dessa lesão.[3][45][46]
A ICC é tratável com diuréticos. A correção cirúrgica envolve fechamento do DSV, separação das circulações pulmonar e sistêmica e colocação de um conduto ventrículo direito-artéria pulmonar. A cirurgia é normalmente realizada por volta dos 2-3 meses de idade para prevenir o desenvolvimento de hipertensão pulmonar com resistência vascular pulmonar elevada. Os pacientes também podem requerer diversas intervenções transcateter ou operações para tratar a disfunção do conduto ventrículo direito-pulmonar.[47] A mortalidade operatória é maior com anomalias associadas, como interrupção do arco aórtico.
Uma malformação rara da valva tricúspide, na qual a delaminação anormal dos folhetos septais e posteriores da valva leva a vários graus de regurgitação tricúspide, que pode ser grave em um estágio inicial da vida.[48][49][50] Como resultado da falta de delaminação e dependendo da sua gravidade, uma parte do ventrículo direito (VD) torna-se "atrializada", o que significa que fica abaixo do nível da valva tricúspide deformada e não contribui para o fluxo anterógrado. Em casos raros, a miopatia do ventrículo esquerdo (VE) também está presente. Em 80% a 94% dos pacientes, está presente comunicação interatrial concomitante.[50] A cianose pode ocorrer devido ao fluxo de sangue desoxigenado do jato de regurgitação da valva tricúspide excêntrica e através da comunicação interatrial.
Os pacientes apresentam a primeira e segunda bulhas cardíacas separadas. A radiografia torácica revela um aumento da área cardíaca significativo com uma borda direita proeminente e vascularidade pulmonar diminuída. O ECG revela ondas "p" altas na derivação II dos membros e um padrão de bloqueio do ramo direito.[50][51] A síndrome de Wolff-Parkinson-White é encontrada em pelo menos 15% dos pacientes. A ecocardiografia, tanto transtorácica quanto transesofágica, auxilia na avaliação das anormalidades estruturais valvares e do VD atrializado.[3] A RNM cardíaca é útil para delinear o grau de atrialização do VD, seu tamanho e sua função.
Há uma faixa ampla de gravidade anatômica e fisiológica. Os pacientes com cardiomegalia e regurgitação tricúspide mínimas levam uma vida normal sem precisar de cirurgia. Os neonatos com formas graves da anomalia de Ebstein requerem prostaglandina E1 para aumentar o fluxo sanguíneo pulmonar até a resistência vascular pulmonar cair, e alguns ficam bem sem cirurgia. Contudo, o controle pode ser difícil em um subconjunto de pacientes com uma cardiomegalia maciça. A regurgitação tricúspide grave é indicação de cirurgia, após cuidadosa avaliação hemodinâmica.[26]
As intervenções cirúrgicas consistem na reparação, substituição ou reparação através da operação com técnica do cone de Da Silva da valva tricúspide, que é uma cirurgia mais extensa em que a valva tricúspide é reparada, o anel é devolvido à sua localização anatômica e o tecido atrial excessivo é removido. Quando realizada por cirurgiões qualificados, os resultados da operação com a técnica do cone em termos de função da valva são excelentes.[52]
Envolvendo <1% de todas as CCs, a CVPAT ocorre quando todas as veias pulmonares se conectam ao sistema venoso sistêmico ou átrio direito. A CVPAT pode ser subdividida em supracardíaca, cardíaca, infracardíaca e tipos mistos, dos quais a supracardíaca é a mais comum. A CVPAT resulta de um sangue altamente oxigenado vindo dos pulmões e sendo direcionado de volta ao átrio direito, e a sobrevida requer uma defeito no septo atrial, sem o qual o sangue não consegue alcançar a aorta.[53] A CVPAT pode ser complicada pela obstrução das veias que drenam a confluência do sistema venoso, resultando em hipertensão pulmonar e congestão.
Os pacientes com veias pulmonares não obstruídas e uma grande comunicação intra-atrial apresentam uma leve cianose e um fluxo sanguíneo pulmonar aumentado. O exame físico revela um impulso ventricular direito proeminente e um sopro sistólico de ejeção. A radiografia torácica revela cardiomegalia moderada e aumento das marcas vasculares pulmonares. A forma supracardíaca de CVPAT revela classicamente uma configuração em "boneco da neve". A ecocardiografia pode mostrar átrio direito e ventrículo direito dilatados e confluência venosa pulmonar posterior.[3]
Os pacientes requerem cirurgia, mas apresentam uma sobrevida em longo prazo excelente.
Os pacientes com fluxo sanguíneo venoso pulmonar obstruído apresentam desconforto respiratório secundário ao edema pulmonar. Isso é comum em neonatos com conexão infracardíaca, e os pacientes precisam de correção cirúrgica emergencial.[53] A mortalidade chega a 40%, apesar da cirurgia imediata.
Resulta na ausência de uma comunicação direta entre o átrio direito e o ventrículo direito. Essa anomalia representa aproximadamente 3% de todas as CCs. A sobrevida inicial é dependente de uma comunicação intra-atrial. A apresentação e o tratamento dependem da relação das grandes artérias (normais ou transpostas), presença de um defeito do septo ventricular (DSV) e gravidade da estenose pulmonar valvar.
Tipicamente, os pacientes apresentam cianose. O impulso apical pode ser proeminente. A segunda bulha cardíaca pode ser única ou desdobrada. Um sopro sistólico pode ser decorrente de DSV ou estenose pulmonar, enquanto um sopro apical mesodiastólico pode ser decorrente do aumento do fluxo sanguíneo pulmonar e da insuficiência cardíaca congestiva.
A radiografia torácica revela cardiomegalia com uma borda atrial direita proeminente. As marcas vasculares pulmonares variam dependendo do grau de estenose pulmonar. O eletrocardiograma (ECG) é caracterizado pelo desvio do eixo esquerdo e uma rotação inicial anti-horária do plano frontal. Pode haver hipertrofia ventricular esquerda.
Se a fonte do fluxo sanguíneo pulmonar não for confiável, o paciente deve ser tratado com prostaglandina E1 até que um desvio circulatório sistêmico-pulmonar possa ser estabelecido. Pacientes com grandes artérias transpostas apresentam um fluxo sanguíneo pulmonar irrestrito e precisam de bandagem arterial pulmonar para evitar a doença vascular pulmonar obstrutiva e/ou edema pulmonar, em conjunto com medicamentos anticongestivos. Todos os pacientes são finalmente submetidos a uma operação de Fontan, na qual o retorno venoso sistêmico é conectado diretamente às artérias pulmonares (conexão cavo-pulmonar total).[38] A sobrevida em longo prazo excede 85% no acompanhamento de 10 anos pós-cirurgia. As complicações em longo prazo da operação de Fontan incluem anomalias de condução, arritmia, distúrbios dos vasos linfáticos que levam a bronquite plástica e enteropatia perdedora de proteínas, hepatopatia congestiva com fibrose hepática e cirrose final e insuficiência da circulação de Fontan que justifica o transplante cardíaco.
Refere-se a um espectro de lesões que inclui septo ventricular intacto com subdesenvolvimento da ventrículo esquerdo e das valvas mitrais e aórticas. Os pacientes comumente apresentam coarctação da aorta. Os neonatos podem parecer normais por vários dias após o nascimento, mas são dependentes da persistência do canal arterial (PCA). Uma vez que o ducto fecha, eles desenvolvem choque.
O exame físico revela pulsos diminuídos nos membros inferiores e um impulso ventricular direito aumentado. A ecocardiografia é utilizada para avaliação pré e pós-operatória, embora a visualização da anatomia da artéria coronária possa ser um desafio devido à hipoplasia aórtica comumente associada.[46]
Quando não tratada, a SCEH é fatal, embora a mortalidade tenha sido significativamente reduzida na última década. Os pacientes requerem uma abordagem cirúrgica em estágios.[54]
O estágio 1 é o procedimento de Norwood que consiste na reconstrução do arco aórtico usando a artéria pulmonar principal e um desvio de Blalock-Taussig modificado fornecendo fluxo sanguíneo aos ramos arteriais pulmonares. Recentemente, a colocação de um desvio circulatório do ventrículo direito para a artéria pulmonar (Sano) tem sido usado como uma alternativa ao desvio de Blalock-Taussig modificado durante a paliação do estágio 1.[54] O estágio 2 consiste na colocação de um desvio de Blalock-Taussig modificado (ou desvio de Sano) com uma anastomose cavo-pulmonar (Glenn) bidirecional. O estágio 3 consiste na finalização da conexão cavo-pulmonar e formação da circulação de Fontan, conforme detalhado na seção sobre atresia tricúspide. Nos últimos anos, procedimentos como cirurgia híbrida/cateterismo intervencionista têm sido usados com bandagens dos ramos das artérias pulmonares para restringir o fluxo sanguíneo pulmonar e colocação de endopróteses (stents) para manter a patência ductal, embora a utilidade dessa abordagem permaneça incerta. O transplante cardíaco é, às vezes, tido como uma estratégia alternativa à cirurgia em estágios.[55] As complicações em longo prazo da circulação de Fontan são semelhantes às descritas na seção sobre atresia tricúspide, e há algumas evidências de que, na presença de um ventrículo direito sistêmico, estas podem ocorrer em um estágio mais precoce.
Inclui 4 subtipos: estenose da valva aórtica, estenose supravalvular, estenose subvalvular distinta e estenose subaórtica em túnel.
A estenose da valva aórtica é a mais comum e representa 5% de todas as cardiopatias congênitas (CCs).[56] Os folhetos ou cúspides geralmente são malformados ou espessos. Aproximadamente 2% da população geral possui uma valva aórtica bicúspide, mas muitos desses indivíduos nunca desenvolvem uma estenose ou regurgitação da valva aórtica clinicamente significativas. A estenose supravalvular ocorre como uma área de estreitamento distinto ou estreitamento difuso apenas distal à junção sinotubular na aorta ascendente. Está frequentemente associada à síndrome de Williams ou a um defeito no gene elastina no cromossomo 7. A estenose subvalvular distinta ocorre quando há uma crista fibromuscular que causa obstrução logo abaixo da valva aórtica. A turbulência do fluxo sanguíneo pode causar dano aos folhetos valvares da valva aórtica, resultando em regurgitação aórtica progressiva. A estenose subaórtica em túnel refere-se a um segmento estenótico mais longo da via de saída quando comparado à estenose subvalvular distinta.
A apresentação depende da gravidade da obstrução. A maioria dos pacientes é assintomática. Os pacientes mais velhos podem apresentar dor torácica ou síncope. Um subconjunto de pacientes apresenta função ventricular esquerda deficiente, débito cardíaco baixo e sinais de choque e insuficiência cardíaca congestiva (estenose aórtica "crítica").
O exame físico revela uma sopro sistólico em crescendo-decrescendo, mais bem auscultado na borda esternal esquerda com radiação para a borda esternal superior direita. A estenose moderada a grave resulta em frêmito palpável e pulsos tardios. Um clique de ejeção também pode ser auscultado logo após a primeira bulha cardíaca em pacientes com estenose da valva aórtica. O clique não varia com a respiração, em contraste com o clique de ejeção da valva pulmonar. Os pacientes com regurgitação aórtica concomitante também apresentam um sopro diastólico em decrescendo e uma ampla pressão de pulso.
O eletrocardiograma (ECG) inconsistentemente revela evidência de hipertrofia ventricular esquerda. A estenose da VSVE grave está associada com o achatamento ou inversão das ondas T nas derivações V5 e V6.
A radiografia torácica pode revelar uma aorta proeminente como um resultado da dilatação pós-estenótica. A ecocardiografia auxilia na avaliação precisa da morfologia aórtica e na estimativa do gradiente de pressão através da via de saída do ventrículo esquerdo (VSVE). Na estenose aórtica valvar, o gradiente médio estimado pelo Doppler é usado para avaliar a gravidade. O gradiente irá variar de acordo com o débito cardíaco.
Os pacientes com estenose aórtica "crítica" precisam de alívio emergencial da estenose. Os pacientes sintomáticos também necessitam de alívio da obstrução, independentemente do grau da estenose. Os lactentes com estenose valvar grave precisam de valvuloplastia por balão, embora a cirurgia possa ser necessária para regurgitações aórticas coexistentes. A valvotomia cirúrgica ou substituição cirúrgica valvar é considerada para crianças mais velhas e adultos. Os pacientes com estenose grave seguidos no Segundo Estudo da História Natural de Defeitos Cardíacos Congênitos (Second Natural History Study of Congenital Heart Defects) tiveram uma taxa de sobrevivência de 25 anos de 81%.[57] A dilatação da aorta, seja ao nível da raiz ou da parte ascendente, está presente em 20% a 30% dos pacientes com valva aórtica bicúspide, e em pacientes adultos deve ser substituída quando atingir diâmetro de 5.5 cm, ou 5 cm se for necessária uma cirurgia da valva aórtica ou se o paciente tiver um aneurisma aórtico torácico hereditário.[58]
Constitui 5% das CCs e é mais comum em homens; a coarctação aórtica está associada com a síndrome de Turner em mulheres. Esta consiste de uma elevação tecidual posterior que se salienta para dentro da aorta e é tipicamente descrita como justaductal porque ocorre através do canal arterial. É comumente associada a uma valva aórtica bicúspide (70% a 75% dos pacientes com coarctação aórtica têm uma valva aórtica bicúspide, mas apenas cerca de 7% dos pacientes com valva aórtica bicúspide também apresentam coarctação aórtica) e menos comumente com um defeito do septo ventricular ou estenose aórtica subvalvar.[59]
Os pacientes com obstrução leve podem não apresentar, até a adolescência, um sopro cardíaco ou hipertensão sistêmica. Nos pacientes com obstrução grave, a persistência do canal arterial (PCA) é a principal fonte de fluxo sanguíneo sistêmico distal à obstrução, o que significa que os neonatos desenvolvem acidose metabólica e choque conforme o ducto fecha.
Os membros inferiores têm menor saturação de oxigênio e pulsos diminuídos, além de insuficiência cardíaca congestiva em pacientes com obstrução grave. É importante aferir a pressão arterial em ambos os membros superiores e em 1 perna antes de se fazer o diagnóstico, uma vez que é fácil errar o diagnóstico em pacientes com coarctação proximal à artéria subclávia ou em aqueles com uma origem anômala da artéria subclávia direita. O fluxo sanguíneo turbulento através da região de coarctação produz um sopro sistólico. Os pacientes com uma valva aórtica bicúspide associada têm um clique de ejeção.
Os lactentes, paradoxalmente, revelam hipertrofia ventricular direita no ECG e ecocardiografia por causa da PCA abastecendo a aorta distal à obstrução. Os pacientes mais velhos apresentam sinais de hipertrofia ventricular esquerda. A TC e a RNM podem ser úteis na definição da anatomia da coarctação no implante de stent. O método de reparo cirúrgico mais comumente utilizado é uma anastomose término-terminal estendida, que quando realizada por operadores qualificados, tem excelente duração e desfechos em longo prazo.[60] Outras técnicas cirúrgicas incluem reparo de retalho subclávio e uso de enxerto protético. O reparo no período neonatal está associado a um aumento da incidência de recorrência, que é melhor tratada com a colocação de endoprótese (stent). Outras complicações em longo prazo incluem formação de aneurisma no local da anastomose e dissecção da aorta. Apesar da sua natureza localizada, sabe-se bem que a coarctação aórtica causa disfunção endotelial sistêmica, e muitos pacientes correm o risco de desenvolver hipertensão de início precoce, o que pode levar a eventos cardiovasculares adversos, como AVC, doença arterial periférica e cardiopatia isquêmica.[61] Até 10% dos pacientes desenvolvem microaneurismas vasculares intracranianos (às vezes chamados de "aneurisma sacular"), especialmente na área anatômica do círculo de Willis.[62] O acompanhamento por toda a vida, mesmo em pacientes que não apresentam obstrução residual, é extremamente importante.
Para obter mais informações, consulte Coarctação aórtica.
Representa até 8% de todas as cardiopatias congênitas (CCs) e é comumente encontrada em pacientes com síndrome de Noonan. A maioria das crianças apresenta um sopro assintomático. Contudo, os neonatos com estenose pulmonar valvar crítica apresentam cianose secundária a um desvio sanguíneo direita-esquerda no nível atrial.[63]
O exame físico revela um impulso ventricular direito aumentado, um clique após a primeira bulha cardíaca que varia com a respiração, um desdobramento normal a amplo da segunda bulha cardíaca dependendo da gravidade, e um sopro de ejeção em crescendo-decrescendo. Com o aumento da gravidade da estenose, o clique de ejeção pulmonar ocorre precocemente na sístole; nos casos mais graves, o clique pode se fundir com a primeira bulha cardíaca e se tornar inaudível.[49] Reciprocamente, a segunda bulha cardíaca desdobra-se mais amplamente com o aumento da gravidade da estenose e pode tornar-se fixa na estenose grave. Na estenose muito grave, o componente pulmonar da segunda bulha pode tornar-se difícil de auscultar devido ao sopro alto que se difunde na diástole. Uma quarta bulha cardíaca pode ser auscultada em pacientes com insuficiência ventricular direita.[49]
O eletrocardiograma (ECG) pode revelar desvio do eixo direito e uma radiografia torácica pode revelar sinais de hipertrofia ventricular direita. Tipicamente, há uma dilatação pós-estenótica das artérias pulmonares.
A valvoplastia por balão é o tratamento de escolha e, no acompanhamento de 10, 84% dos pacientes não necessitam de intervenção adicional.[63] O desfecho em longo prazo é excelente. Se a substituição da valva for necessária, ela poderá ser realizada cirurgicamente ou por meio de uma abordagem transcateter.
Para obter mais informações, consulte Estenose pulmonar.
As pessoas com CC podem apresentar aumento do risco de infecção por COVID-19 mais grave, particularmente aquelas com características anatômicas e fisiológicas mais graves da CC.[64][65] Os defeitos biventriculares simples, os defeitos ventriculares complexos e um histórico de cirurgia cardíaca estão associados a COVID-19 grave.[66] Foram publicadas recomendações para a prevenção e o manejo da COVID-19 em adultos com doença cardíaca congênita, com base na estratificação do risco.[65][67]
Recomenda-se que os medicamentos cardíacos, incluindo a aspirina, os inibidores da ECA (IECAs), antagonistas do receptor de angiotensina II, betabloqueadores, diuréticos e medicamentos antiarrítmicos sejam mantidos durante a COVID-19, a menos que haja uma contraindicação clara.[64]
Para obter mais informações, consulte Doença do coronavírus 2019 (COVID-19).
O uso deste conteúdo está sujeito ao nosso aviso legal