Datos
La cardiopatía congénita (CC) es el defecto congénito más frecuente, aunque sigue siendo relativamente raro. Centers for Disease Control and Prevention: facts about congenital heart defects Opens in new window El cribado para detectar la cardiopatía coronaria fetal incluye la ecografía en el segundo trimestre del embarazo y la exploración clínica posnatal; no obstante, las tasas de detección son reducidas.[1][2][3] La ecocardiografía fetal se está convirtiendo en la prueba definitiva para el diagnóstico. La Sociedad Americana de Ecocardiografía (American Society of Echocardiography) ha elaborado recomendaciones sobre cuándo derivar para la obtención de imágenes fetales, aunque reconoce que existen diferencias locales en las indicaciones de derivación en función de la población que se someta a las pruebas de detección.[4]
Resulta importante reconocer la CPC de manera temprana porque la presentación clínica y el deterioro pueden ser repentinos y algunos defectos tratables incluso pueden producir la muerte antes de que se obtenga el diagnóstico.[1]
Los tratamientos quirúrgicos y médicos de la CPC han mejorado significativamente durante los últimos 50 años. Las primeras cirugías exitosas a corazón abierto para defectos intracardíacos se llevaron a cabo en los Estados Unidos en la década de 1950.[5][6][7] La introducción de máquinas que perfundían los órganos vitales mientras un cirujano reparaba cuidadosamente un corazón parado supuso una revolución en el campo de la cirugía correctiva. Actualmente se espera que la mayoría de los bebés que nacen con CPC logren sobrevivir hasta la edad adulta.[8]
En todo el mundo, se estima que la prevalencia de las cardiopatías congénitas (CPC) es del 0.8% al 1.2% de los nacidos vivos.[9] En la América del Norte de ingresos altos, incluidos los EE. UU., se estima que la prevalencia de CPC es de 12.3 por 1000 (IC del 95%, 10.9 a 13.8). Se estima que un 1% o un mínimo de 40,000 bebés por año se verán afectados por la CPC cada año en los EE. UU. Alrededor del 25%, o 2.4 por cada 1000 nacidos vivos, se someterán a un tratamiento invasivo en el primer año de vida. El tipo más común de cardiopatía congénita es la válvula aórtica bicúspide, que se presenta en 13.7 de cada 1000 personas.[10]
Las anomalías cromosómicas (por ejemplo, el síndrome de DiGeorge, el síndrome de Down o el síndrome de Turner) se encuentran en el 8% al 10% de las personas con cardiopatías coronarias, y las variantes de un solo nucleótido o del número de copias patógenas se encuentran en el 5% al 15%.[10]
Informes recientes muestran una disminución significativa de la mortalidad entre los pacientes con CPC. En 2020, hubo 2817 muertes relacionadas con la CPC en los EE. UU., una disminución del 11.9% con respecto a 2010, y la tasa de mortalidad ajustada por edad fue de 0.9 muertes por cada 100,000 personas, una disminución del 18.2% con respecto a 2010.[10] Debido a la mejora de la supervivencia, ahora hay una población envejecida de adultos con CPC que presentan complicaciones relacionadas con la enfermedad cardiovascular adquirida (p. ej., eventos tromboembólicos y arritmias).[8][11][12] El aumento del riesgo de morbilidad y mortalidad por enfermedad cardiovascular aterosclerótica (ASCVD, por sus siglas en inglés) prematura en este grupo plantea un desafío cada vez mayor y destaca la necesidad de un seguimiento a largo plazo de los pacientes y el conocimiento de sus factores de riesgo de ASCVD.[8]
Derivaciones de izquierda a derecha
Lesiones que permiten que la sangre derive del lado izquierdo del corazón al derecho. Se asocian a diversos grados de aumento del flujo sanguíneo pulmonar y normalmente son acianóticas. En algunos defectos, el sitio de la derivación puede no ubicarse en el propio corazón.
La cianosis se produce solo si las lesiones son grandes y no se reparan en la niñez, y si el paciente desarrolla una enfermedad vascular pulmonar obstructiva (fisiología de Eisenmenger). La ecocardiografía es la modalidad de imagenología primaria y, en la época actual, el papel del cateterismo cardíaco se centra principalmente en la intervención.[13]
Por ejemplo,
Comunicación interventricular (CIV)
Comunicación interauricular (CIA)
Comunicación auriculoventricular (CAV)
Conducto arterioso permeable (CAP)
Conexión venosa pulmonar anómala parcial (CVPAP).
Derivaciones de derecha a izquierda
Lesiones que provocan que sangre desoxigenada llegue a la aorta y se asocian a un aumento o disminución del flujo sanguíneo pulmonar.
Por ejemplo,
Tetralogía de Fallot (TF)
Atresia de válvula pulmonar con o sin una comunicación interventricular (CIV)
Dextro-transposición de las grandes arterias (d-TGA)
Tronco arterioso
Anomalía de Ebstein
Conexión venosa pulmonar anómala total (CVPAT)
Síndrome del ventrículo izquierdo hipoplásico (SVIH).
Lesiones valvulares y no valvulares obstructivas
Obstrucción del tracto de salida ventricular izquierdo (TSVI)
Coartación aórtica
Estenosis de la válvula pulmonar (EVP)
Estenosis de la válvula aórtica (EVA)
Los defectos cardíacos congénitos también se pueden clasificar utilizando la clasificación anatómica/fisiológica (AP) introducida por el grupo de trabajo de la American Heart Association/American College of Cardiology (AHA/ACC) en sus guías de práctica clínica de 2018.[14]
En el sistema AP, un paciente se clasifica con una clase "A" y una clase "P" en función de la característica anatómica o fisiológica relevante "más alta", y los pacientes pueden pasar de una clasificación AP a otra con el tiempo. Si el estado clínico empeora, la clasificación cambiará a un grupo de gravedad superior, pero la mejoría del estado; por ejemplo, tras una intervención como la sustitución valvular o el control de la arritmia, puede dar lugar a un cambio a una clasificación de gravedad inferior.
En las guías de práctica clínica de 2020 para el manejo de adultos con cardiopatías congénitas publicadas por la Sociedad Europea de Cardiología (ESC), solo se utiliza la clasificación anatómica, sin el componente fisiológico.[15] Existen diferencias en la clasificación de gravedad en ambos sistemas de clasificación; por ejemplo, la estenosis congénita de la válvula aórtica y la enfermedad congénita de la válvula mitral se consideran defectos leves en las guías de práctica clínica de la ESC, mientras que las guías de práctica clínica de la AHA/ACC las clasifican como moderadas.
La CIV la forma más frecuente de cardiopatía congénita (CPC) al representar el 20% de todos los casos, sin incluir la válvula aórtica bicúspide y el prolapso de la válvula mitral.[16] Los subtipos que se basan en la ubicación incluyen: perimembranosa (en la zona del tabique membranoso); de salida (debajo de 1 o ambas válvulas semilunares); de entrada (inferiores a las válvulas auriculoventriculares [AV]); y muscular (en la parte muscular o trabeculada del tabique ventricular).[17][Figure caption and citation for the preceding image starts]: Los subtipos de las comunicaciones interventriculares son: (A) salida; (B) perimembranosa; (C) entrada; (D) muscularMayo Clinic Foundation [Citation ends].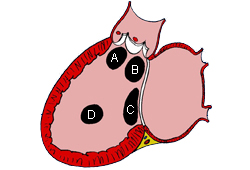
Los defectos perimembranosos suelen presentarse como una lesión solitaria y, en ocasiones, pueden cerrarse espontáneamente por aposición de la valva del tabique de la válvula tricúspide en el defecto. Los defectos de salida pueden ser grandes y estar asociados a formas más complejas de cardiopatía congénita, como la tetralogía de Fallot. Tanto las CIV perimembranosas como las de salida se encuentran muy cerca de la valva semilunar derecha de la válvula aórtica. Debido al efecto Venturi, estos defectos pueden provocar prolapso de una valva de la válvula aórtica, lo que produce restricción en el flujo a través de la CIV y regurgitación valvular aórtica.[18] Los defectos de entrada no se cierran espontáneamente y pueden estar asociados a CAV e insuficiencia valvular AV. Los defectos musculares son el tipo más frecuente de CIV en recién nacidos y la gran mayoría se cierran espontáneamente antes de los 2 años de edad.[Figure caption and citation for the preceding image starts]: Imagen de ecocardiografía de 4 cámaras apical de una comunicación interventricular (CIV) muscular (flecha). (AD) aurícula derecha; (AI) aurícula izquierda; (VD) ventrículo derecho; (VI) ventrículo izquierdoImagen cortesía de Patrick W. O'Leary, MD [Citation ends].
Los niños con una CIV pequeña se desarrollan normalmente y son asintomáticos. Con defectos más grandes, el exceso de flujo sanguíneo pulmonar que se produce después de las 6-8 semanas de edad, cuando se produce la disminución normal del flujo sanguíneo pulmonar, puede conducir al desarrollo de taquipnea, taquicardia, palidez, mala alimentación y poco aumento de peso. El remodelado vascular pulmonar y la enfermedad obstructiva generalmente no se desarrollan en lactantes, pero puede desarrollarse a los 2 años de edad. Si no se trata, la mayoría de las CIV de moderadas a grandes eventualmente conducen a hipertensión pulmonar grave y, en algún momento, se produce la reversión de la derivación, lo que conduce a cianosis central. Esta afección, llamada síndrome de Eisenmenger, no suele producirse hasta más tarde en la vida. Una vez que un paciente desarrolla la fisiología de Eisenmenger, el cierre de la CIV está contraindicado.[19][20]
Los pacientes presentan un soplo holosistólico de frecuencia baja a media, un impulso precordial prominente y, en pacientes con hipertensión pulmonar, un segundo ruido cardíaco único y fuerte. Si la derivación de izquierda a derecha es grande, los pacientes desarrollan un soplo diastólico medio en el flujo ("ruido") a lo largo de la válvula mitral.
La radiografía de tórax (RT) y el electrocardiograma (ECG) pueden ser normales en las CIV pequeñas. En defectos más grandes, hay un agrandamiento del corazón y aumento de las tramas vasculares pulmonares en la RT y el ECG revela una hipertrofia ventricular izquierda; la hipertrofia ventricular derecha se puede producir con defectos más grandes. Las CIV de entrada se asocian a una desviación del eje izquierdo en el ECG. La ecocardiografía proporciona información importante relativa a la anatomía del defecto, el volumen de la derivación y la presión ventricular derecha.[21][22][Figure caption and citation for the preceding image starts]: RT que demuestra una circulación pulmonar excesivaMayo Clinic Foundation [Citation ends].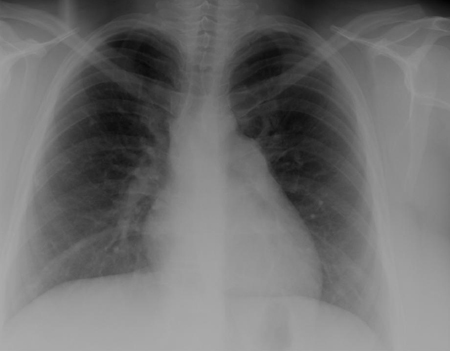
El objetivo del tratamiento es garantizar el crecimiento somático adecuado y evitar la enfermedad vascular pulmonar obstructiva. En ausencia de desarrollo de hipertensión pulmonar, las CIV suelen conducir a un agrandamiento del ventrículo izquierdo (VI), que sirve como indicación para el cierre. Las CIV pequeñas con flujo restrictivo pueden dejarse sin tratar y es necesario realizar una vigilancia continua para detectar el desarrollo de agrandamiento del VI y endocarditis. Las CIV grandes y los defectos asociados a insuficiencia valvular aórtica debido a un prolapso de una valva de la válvula aórtica o a flujo sanguíneo pulmonar excesivo pueden requerir diuréticos y un aumento de la fórmula calórica, seguido del cierre del defecto (ya sea un cierre quirúrgico o transcatéter). Aquellas que presenten un aumento del flujo sanguíneo pulmonar y retraso en el crecimiento se cierran quirúrgicamente entre los 1 y 4 meses de edad; los defectos más grandes se cierran normalmente entre los 6 y 9 meses de edad. Para los pacientes con síndrome de Eisenmenger, la terapia vasodilatadora pulmonar es el tratamiento de elección, y se debe informar a las mujeres en edad fértil que no se recomienda el embarazo debido a la alta mortalidad materna y las complicaciones fetales.[19][20]
Para obtener más información, consulte Comunicación interventricular.
Las comunicaciones interauriculares incluyen comunicación interauricular (CIA) y otras derivaciones pretricuspídeas (p. ej., defecto del seno venoso y seno coronario destechado). Las derivaciones pretricuspídeas se definen como la presencia de una derivación de izquierda a derecha que se produce proximal a la válvula auriculoventricular derecha. En comparación con las derivaciones post-tricuspídeas (como la comunicación interventricular y el conducto arterioso permeable), estos defectos se caracterizan por una derivación de baja presión y un grado variable de derivación de volumen. Las derivaciones pretricuspídeas representan entre el 6% y el 10% de todas las CPC y tienen un predominio de mujeres a hombres de 2:1.[23] Los principales tipos consisten en la comunicación interauricular tipo ostium secundum (60% a 70% de todas las derivaciones pretricuspídeas), que se producen en la zona de la fosa oval; defecto de tipo ostium primum, una forma de comunicación auriculoventricular (CAV), en la cara inferior del tabique auricular; defectos del seno venoso superior e inferior (en los que el tabique interauricular está intacto), que están presentes en el sitio de entrada de la vena cava superior e inferior, respectivamente, en la aurícula derecha; seno coronario destechado, el tipo menos frecuente.[Figure caption and citation for the preceding image starts]: Subtipos de comunicaciones interauriculares: (A) seno venoso; (B) ostium secundum; (C) ostium primum; (D) seno coronario sin techoMayo Clinic Foundation [Citation ends].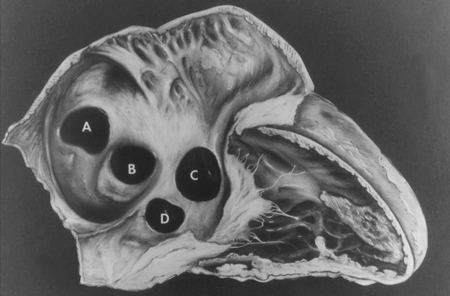 [Figure caption and citation for the preceding image starts]: Imagen de ecocardiografía de 4 cámaras apical de una comunicación interauricular (CIA) de tipo ostium primum (flecha). (AD) aurícula derecha; (AI) aurícula izquierda; (VI) ventrículo izquierdoImagen cortesía de Patrick W. O'Leary, MD [Citation ends].
[Figure caption and citation for the preceding image starts]: Imagen de ecocardiografía de 4 cámaras apical de una comunicación interauricular (CIA) de tipo ostium primum (flecha). (AD) aurícula derecha; (AI) aurícula izquierda; (VI) ventrículo izquierdoImagen cortesía de Patrick W. O'Leary, MD [Citation ends].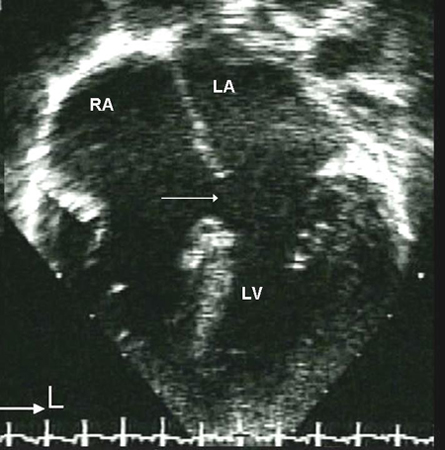
El seno coronario destechado no es en realidad una comunicación interauricular. Se produce cuando el proceso de techado del seno coronario en la vida fetal es incompleto, lo que permite que el seno coronario y la aurícula izquierda se comuniquen.
Los niños con derivaciones pretricuspídeas aisladas suelen ser asintomáticos.[24] Se trata del diagnóstico de CPC que menos se detecta en la infancia y con frecuencia no se descubre hasta la edad adulta. Los defectos no tratados de tamaño moderado o grande conducen a intolerancia al ejercicio, arritmias auriculares y aumento del flujo sanguíneo pulmonar. Un aumento real de la resistencia vascular pulmonar es mucho más raro que en las derivaciones post-tricúspides y se produce más tarde en la vida, más comúnmente en subpoblaciones específicas como las que tienen síndrome de Down. La embolización paradójica a través del defecto se produce casi exclusivamente en pacientes con CIA tipo ostium secundum.
En pacientes con defectos moderados a grandes, el examen revela un aumento del impulso ventricular derecho, un segundo ruido cardíaco fijo ampliamente dividido y un soplo sistólico eyectivo suave que se escucha mejor en el borde esternal superior izquierdo, que es causado por un aumento del flujo sanguíneo a través de la válvula pulmonar, más que a través de la comunicación interauricular. En defectos grandes, también puede estar presente un soplo diastólico en el borde esternal inferior derivado de un aumento del flujo sanguíneo a través de la válvula tricúspide.[24]
La RT no resulta útil para determinar el subtipo y puede ser normal con una CIA pequeña. El ECG también puede ser normal en CIA pequeñas de tipo ostium secundum, de seno venoso y de seno coronario sin techo. Con una derivación más grande, puede haber agrandamiento de la aurícula derecha, hipertrofia ventricular derecha o desviación del eje derecho.[Figure caption and citation for the preceding image starts]: Imagen de ecocardiografía de eje corto paraesternal que demuestra agrandamiento ventricular derecho en un paciente con una comunicación interauricular (CIA). (VD) ventrículo derecho; (VI) ventrículo izquierdoImagen cortesía de Patrick W. O'Leary, MD [Citation ends].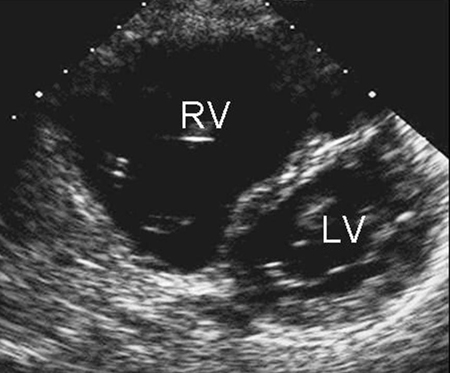 [Figure caption and citation for the preceding image starts]: Imagen de ecocardiografía de 4 cámaras apical que demuestra agrandamiento ventricular derecho en un paciente con una comunicación interauricular (CIA). (AD) aurícula derecha; (VD) ventrículo derecho; (VI) ventrículo izquierdoImagen cortesía de Patrick W. O'Leary, MD [Citation ends].
[Figure caption and citation for the preceding image starts]: Imagen de ecocardiografía de 4 cámaras apical que demuestra agrandamiento ventricular derecho en un paciente con una comunicación interauricular (CIA). (AD) aurícula derecha; (VD) ventrículo derecho; (VI) ventrículo izquierdoImagen cortesía de Patrick W. O'Leary, MD [Citation ends].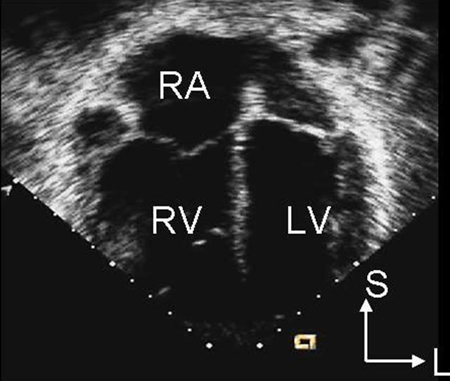 Los defectos de tipo ostium primum se caracterizan por un asa inicial del plano central girada a la izquierda y desviación del eje izquierdo.[24]
Los defectos de tipo ostium primum se caracterizan por un asa inicial del plano central girada a la izquierda y desviación del eje izquierdo.[24]
El tratamiento implica una operación o cierre percutáneo.[25][Figure caption and citation for the preceding image starts]: Imagen de ecocardiografía transesofágica de un dispositivo oclusor de comunicación interauricular (CIA) (flecha). (AD) aurícula derecha; (AI) aurícula izquierda; (VCS) vena cava superiorImagen cortesía de Patrick W. O'Leary, MD [Citation ends].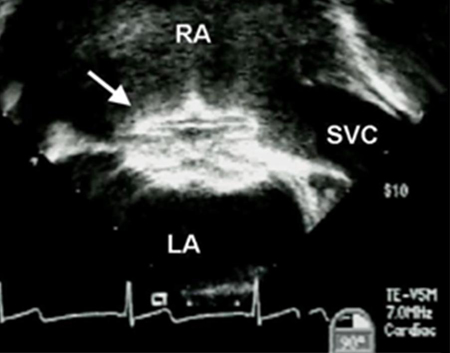 El cierre del dispositivo es el método de elección para los defectos del tipo ostium secundum si el defecto es de un tamaño adecuado (el dispositivo de cierre más grande disponible tiene 40 mm de diámetro) y si hay bordes septales adecuados para asegurar el dispositivo.[26] En la mayoría de los otros subtipos, el cierre del dispositivo no es factible debido a la falta de bordes y la proximidad a otras estructuras cardíacas, aunque informes recientes sugieren el cierre exitoso del dispositivo de defectos del seno venoso superior anatómicamente ajustados.[27] La CAV parcial (CIA de tipo ostium primum y válvula mitral hendida) se puede reparar entre los 18 y 24 meses de edad en la mayoría de los pacientes e implica el cierre de la hendidura mitral además de la CIA.
El cierre del dispositivo es el método de elección para los defectos del tipo ostium secundum si el defecto es de un tamaño adecuado (el dispositivo de cierre más grande disponible tiene 40 mm de diámetro) y si hay bordes septales adecuados para asegurar el dispositivo.[26] En la mayoría de los otros subtipos, el cierre del dispositivo no es factible debido a la falta de bordes y la proximidad a otras estructuras cardíacas, aunque informes recientes sugieren el cierre exitoso del dispositivo de defectos del seno venoso superior anatómicamente ajustados.[27] La CAV parcial (CIA de tipo ostium primum y válvula mitral hendida) se puede reparar entre los 18 y 24 meses de edad en la mayoría de los pacientes e implica el cierre de la hendidura mitral además de la CIA.
Para obtener más información, consulte Comunicaciones interauriculares.
Representa del 4% al 5% de todas las cardiopatías congénitas (CPC) y se detecta en el 40% de los niños con síndrome de Down.[28] Este defecto también se denomina defecto del relieve endocárdico o defecto del canal auriculoventricular (AV). Los relieves endocárdicos cierran la CIA de tipo ostium primum y forman porciones de las válvulas AV y del tabique ventricular. Las CAV pueden ser parciales o completas; un tipo de CAV parcial es una comunicación interauricular (CIA) de tipo ostium primum. Una CAV completa consta de una CIA de tipo ostium primum y una comunicación interventricular (CIV) de entrada contigua.[Figure caption and citation for the preceding image starts]: Imagen de ecocardiografía de 4 cámaras apical de una comunicación auriculoventricular (CAV) completa. Observe la comunicación interauricular (CIA) de tipo ostium primum (*) y la comunicación interventricular (CIV) de entrada contigua (flecha). (AD) aurícula derecha; (AI) aurícula izquierda; (VD) ventrículo derecho; (VI) ventrículo izquierdoImagen cortesía de Patrick W. O'Leary, MD [Citation ends].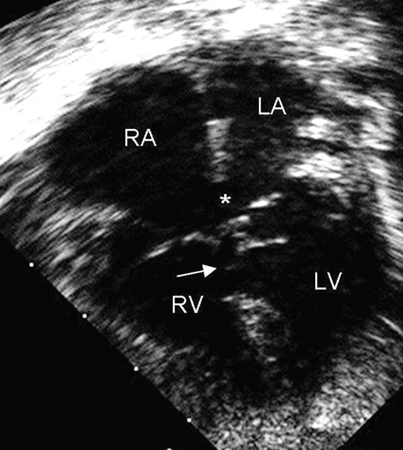
La presentación y hallazgos en la exploración de pacientes con CAV completa y parcial son parecidos a los de pacientes con CIV o CIA, respectivamente. La gravedad también se determina mediante el volumen de la derivación, la extensión de las alteraciones de la válvula AV, anomalías cardíacas y extracardíacas asociadas, y el tamaño relativo de los 2 ventrículos.
El electrocardiograma (ECG) es probable que revele un asa inicial del plano frontal girada a la izquierda y un eje eléctrico del complejo QRS superior.[Figure caption and citation for the preceding image starts]: Electrocardiograma (ECG) de 12 derivaciones en un lactante con comunicación auriculoventricular (CAV) completa; el ECG es significativo para la desviación del eje izquierdoMayo Clinic Foundation [Citation ends].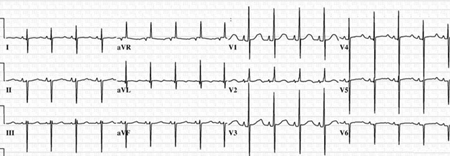 Los pacientes con una CAV completa generalmente tienen cardiomegalia y aumento de las tramas vasculares pulmonares en la RT.[29]
Los pacientes con una CAV completa generalmente tienen cardiomegalia y aumento de las tramas vasculares pulmonares en la RT.[29]
Los niños con una CAV completa requieren reparación con cirugía, generalmente realizada entre los 3 y 6 meses de edad. Estos defectos requieren un seguimiento durante toda la vida, ya que aproximadamente el 15% de los pacientes desarrolla una regurgitación valvular AV progresiva u obstrucción del tracto de salida ventricular izquierdo.[14]
Un conducto arterioso normalmente se cierra unos días después del nacimiento y es un componente esencial de la normalidad en la fisiología cardiovascular del útero. La permeabilidad persistente se produce en 1 de 2000 nacidos vivos en lactantes nacidos a término pero es mucho más frecuente en neonatos prematuros.[30] En lactantes prematuros, se ha observado un CAP durante la admisión al hospital en el 42% de los neonatos con un peso de 500-999 g, el 21% con un peso de 1000-1499 g, y el 7% con un peso de 1500-1750 g.[31] También hay una incidencia 30 veces mayor en pacientes nacidos a altitudes más elevadas. El CAP representa entre el 9% y el 12% de todas las cardiopatías congénitas.
Los pacientes son generalmente asintomáticos cuando el conducto es pequeño. Con el aumento de tamaño, los recién nacidos presentan signos de aumento del flujo sanguíneo pulmonar, presión de pulso amplia y pulso acelerado. La fisiología de Eisenmenger, secundaria a la resistencia vascular pulmonar elevada y a la reversión de la derivación, puede producirse si el CAP es grande y de larga duración, e inicialmente resulta en cianosis solo en la mitad inferior del cuerpo con un aumento gradual de la mezcla sistémica y la cianosis a medida que avanza la enfermedad.[20]
Normalmente, un CAP produce un soplo continuo, que se oye mejor en la zona infraclavicular izquierda. El soplo es continuo porque la presión aórtica es superior a la presión de la arteria pulmonar durante la sístole y la diástole. Si la derivación es grande, también se puede oír un soplo en el flujo mitral diastólico. En algunos lactantes prematuros y recién nacidos el soplo se puede oír únicamente en la sístole y se puede confundir con el soplo de una comunicación interventricular (CIV).
El electrocardiograma (ECG) y la radiografía de tórax (RT) son con frecuencia normales. En un CAP grande, pueden estar presentes hipertrofia biventricular en el ECG de superficie, y aumento del flujo sanguíneo pulmonar y cardiomegalia en la RT. La ecocardiografía permite la delineación de la anatomía del CAP y conocer la dirección y el volumen de la derivación.[32] Se puede obtener una evaluación adicional de la anatomía y la proximidad a otras estructuras con un laboratorio transversal, como un estudio de tomografía computarizada.[22]
En lactantes prematuros, el conducto puede cerrarse con frecuencia con indometacina o ibuprofeno, o si esto no funciona, ligarse quirúrgicamente.[33] En niños mayores y adultos, en ausencia de hipertensión arterial pulmonar significativa, los CAP se cierran percutáneamente mediante espirales o dispositivos.[14][26] Se pueden usar diuréticos para tratar a pacientes con flujo sanguíneo pulmonar excesivo hasta el cierre definitivo.
Para más información consulte Conducto arterioso persistente.
Una anomalía en la que ≥1 (pero no todas) las venas pulmonares se conectan a las venas sistémicas o a la aurícula derecha en lugar de hacerlo a la aurícula izquierda. Este defecto representa <1% de todas las cardiopatías congénitas. La conexión anómala de la(s) vena(s) pulmonar(es) superior/media derecha(s) se asocia con un defecto del seno venoso; la conexión de la(s) vena(s) pulmonar(es) inferior(es) derecha(s) a la vena cava inferior puede formar parte de un defecto del seno venoso inferior, también conocido como "síndrome de cimitarra".[34]
Los hallazgos de presentación, electrocardiograma (ECG) y radiografía de tórax (RT) son parecidos a los de pacientes con CIA, aunque los pacientes con una sola vena anormal suelen ser clínicamente asintomáticos. La presentación clínica de los pacientes con síndrome de cimitarra varía desde adultos relativamente asintomáticos hasta lactantes gravemente enfermos con hipoplasia pulmonar ipsilateral, infecciones de las vías respiratorias e hipertensión pulmonar.[35]
La ecocardiografía transtorácica puede no ser suficiente para diagnosticar CVPAP de manera precisa en adultos, y es posible que sea necesario emplear otras modalidades de imagenología como la ecocardiografía transesofágica, tomografía computarizada (TC) o imagen por resonancia magnética (IRM).
La intervención quirúrgica se indica si el volumen de la derivación crea una sobrecarga de volumen significativa en el lado derecho, que se asocia a un soplo en el tracto de salida pulmonar y ocasionalmente a un soplo tricuspídeo diastólico en el flujo de entrada.[24]
La TOF es la cardiopatía coronaria cianótica más frecuente, que representa entre el 4% y el 8% de todos los defectos, y puede producirse como una lesión cardíaca solitaria o como parte de un síndrome genético, el más frecuente de los cuales es el síndrome de DiGeorge (una microdeleción de 22q11.2).[36] Consiste en 4 anormalidades: comunicación interventricular (CIV) de salida, obstrucción del tracto de salida ventricular derecho (TSVD), cabalgamiento de la aorta en la cresta del tabique ventricular, e hipertrofia ventricular derecha (VD), resultado de una obstrucción del TSVD.[Figure caption and citation for the preceding image starts]: Imagen de ecocardiografía de eje largo paraesternal en un paciente con tetralogía de Fallot. Hay cabalgamiento de la aorta sobre la comunicación interventricular (*). (AI) aurícula izquierda; (VD) ventrículo derecho; (VI) ventrículo izquierdoImagen cortesía de Patrick W. O'Leary, MD [Citation ends].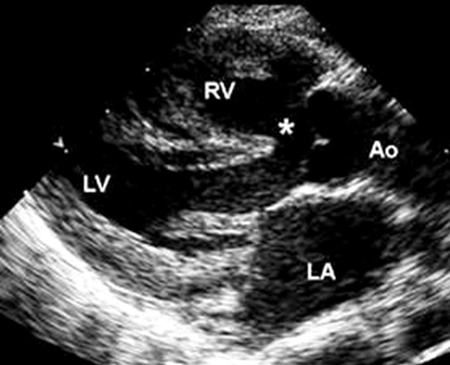
Si la obstrucción del TSVD es leve, se produce una derivación leve o nula de derecha a izquierda y no hay cianosis aparente, un entorno fisiológico que a veces se denomina "tetralogía rosa". No obstante, la mayoría de los pacientes con TF tiene cianosis progresiva tras el nacimiento seguida de disnea de esfuerzo cuando son niños pequeños. Se desarrolla una eritrocitosis secundaria como mecanismo de compensación de la hipoxemia crónica. Puede presentarse un crecimiento somático deficiente. La hipercianosis o episodios cianóticos son episodios hipóxicos que consisten en una aparición repentina de respiración rápida y superficial, mayor agitación, cianosis y un descenso de la intensidad del soplo debido a una reducción del flujo sanguíneo por el tracto de salida ventricular derecho. Sin tratamiento, estos episodios pueden causar hipoxemia profunda, convulsiones y la muerte. Los niños mayores con episodios hipóxicos se pondrán de cuclillas para aumentar la resistencia vascular sistémica y, de esta forma, reducir la hipoxemia.
Se oye un soplo sistólico eyectivo fuerte en el borde esternal superior izquierdo con radiación a ambas axilas debido a la obstrucción del tracto de salida ventricular derecho. En muy raras ocasiones, los pacientes con una obstrucción mínima del tracto de salida ventricular derecho presentan solo una cianosis leve y pueden desarrollar signos de flujo sanguíneo pulmonar excesivo debido a una derivación de izquierda a derecha por la CIV. Puede estar presente un impulso ventricular derecho.
De forma clásica, la radiografía de tórax (RT) es normal excepto cuando hay una disminución de las tramas vasculares pulmonares. Menos del 25% de los lactantes tiene la silueta cardíaca descrita clásicamente como "en forma de bota".
Los lactantes con hipoxia grave se pueden tratar con prostaglandina E1 para volver a abrir o mantener la permeabilidad del conducto arterioso y aumentar el flujo sanguíneo pulmonar. Si este tratamiento no mejora la hipoxemia, el paciente puede requerir un injerto sistémico-pulmonar, como una derivación Blalock-Taussig modificada, hasta que la reparación sea definitiva (que incluye el cierre con parche de la CIV y el alivio de la obstrucción del tracto de salida ventricular derecho).[37] La reparación completa se puede realizar en ciertos centros en el periodo neonatal, aunque la mayoría de los médicos prefiere realizarla a los 3 meses de edad o cuando se desarrolle una cianosis progresiva. Los resultados quirúrgicos han sido excelentes en las últimas décadas, aunque muchos pacientes "reparados" cuando eran niños pequeños requieren la reimplantación de una válvula pulmonar competente cuando son mayores, lo que puede realizarse quirúrgicamente o en un procedimiento transcatéter.[14]
El laboratorio de seguimiento es esencial para identificar la necesidad oportuna de reemplazo valvular, el tipo de tratamiento requerido y para monitorizar el resultado del tratamiento.[38] Las arritmias ventriculares son una complicación potencial a largo plazo que pone en riesgo la vida de la TF reparada, y la etiología probable es la conducción lenta de los istmos anatómicos creados en el momento de la reparación quirúrgica original. El tratamiento incluye la ablación con o sin desfibrilador cardioversor intracardíaco.[39]
Para más información consulte Tetralogía de Fallot.
Con frecuencia se considera la forma más grave de tetralogía de Fallot en la que no se desarrolla la válvula pulmonar. El setenta por ciento de los pacientes tiene un conducto arterioso permeable (CAP) que mantiene el flujo sanguíneo pulmonar. El resto tiene varios vasos colaterales sistémico-pulmonares. Las anomalías de la arteria pulmonar son frecuentes e incluyen hipoplasia, son no confluentes y presentan una distribución anormal.[40]
Los pacientes son generalmente cianóticos de nacimiento. Puede que sea audible un soplo continuo del CAP o de los colaterales sistémico-pulmonares. El segundo ruido cardíaco es único. El electrocardiograma (ECG) muestra una desviación del eje derecho e hipertrofia ventricular derecha. La radiografía de tórax (RT) revela un descenso notable en la vascularidad pulmonar.
Se comienza con prostaglandina E1 para mantener la permeabilidad del conducto arterioso. Es también probable que la mayoría de los pacientes requiera una desviación sistémica-pulmonar para mantener el flujo sanguíneo pulmonar adecuado. Los pacientes con grandes arterias pulmonares confluentes pueden someterse a una reparación de una sola etapa; el resto requiere una reparación de varias etapas. Los pacientes con múltiples colaterales de la arteria sistémica a pulmonar tienen el riesgo de desarrollar hipertensión pulmonar temprana, que inicialmente puede ser segmentaria.[20]
Este defecto es raro y su gravedad depende de la hipoplasia de la válvula tricúspide y del ventrículo derecho asociada con frecuencia. Las fistulas de la arteria coronaria al ventrículo derecho también se asocian a este defecto.[41] Es necesaria una comunicación interauricular (CIA) grande y sin restricción para sobrevivir ya que no hay salida del ventrículo derecho a las arterias pulmonares.
No hay hallazgos del examen característicos de este defecto. La radiografía de tórax (RT) revela cardiomegalia con una aurícula derecha prominente y el electrocardiograma (ECG) muestra un agrandamiento de la aurícula. La ecocardiografía se utiliza para determinar el tamaño ventricular derecho y la hipoplasia de la válvula tricúspide.
Una CIA con restricción es una emergencia médica en estos pacientes y requiere una septostomía auricular con balón urgente. Los pacientes se deben tratar con prostaglandina E1 para mantener la permeabilidad ductal porque el conducto arterioso permeable es la única fuente del flujo sanguíneo pulmonar.[42][43]
La mayoría de los pacientes requiere operaciones por etapas que potencialmente permiten que la válvula tricúspide y el ventrículo derecho aumenten de tamaño. Puede ser necesaria una operación de Fontan modificada o una reparación de 1.5 ventrículos si el ventrículo derecho y la válvula tricúspide no alcanzan un tamaño adecuado para permitir una reparación de 2 ventrículos. Una reparación de 1.5 ventrículos implica una derivación cavopulmonar bidireccional (anastomosis 'Glenn') para desviar el flujo sanguíneo de la vena cava superior a las arterias pulmonares y así reducir la precarga ventricular derecha, el cerrar la CIA y el establecer la continuidad del ventrículo derecho a la arteria pulmonar. Los pacientes con fístulas de la arteria coronaria al ventrículo derecho pueden requerir una operación de Fontan modificada.
Cardiopatía congénita (CPC) común que representa el 4% de todos los defectos. En este defecto, la aorta asciende desde el ventrículo derecho y la arteria pulmonar asciende desde el ventrículo izquierdo. Como resultado, la sangre venosa sistémica desoxigenada viaja desde el ventrículo derecho hasta la aorta sin pasar por los pulmones, y la sangre venosa pulmonar viaja desde el ventrículo izquierdo hasta los pulmones por la arteria pulmonar. Las circulaciones sistémicas y pulmonares están en paralelo en lugar de en serie. Esto es incompatible con la vida sin una comunicación entre los 2 circuitos, como una comunicación interventricular (CIV), comunicación interauricular (CIA) o conducto arterioso permeable (CAP). La mayoría de los pacientes con d-TGA no tiene una CIV y un tercio de los que tienen una CIV tiene estenosis pulmonar (EP) asociada.
Los pacientes presentan cianosis en las primeras horas de nacimiento como emergencia médica.
El examen revela un impulso ventricular derecho prominente, un único segundo ruido cardíaco fuerte (debido a la posición anterior de la aorta) y ningún soplo, o un soplo sistólico eyectivo en aquellos con una CIV y estenosis pulmonar.
Se utiliza prostaglandina E1 para mantener la permeabilidad del conducto arterioso y la mayoría de los pacientes también requiere una septostomía auricular para mejorar la mezcla en el nivel auricular. La operación de cambio arterial, que incluye el reimplante del botón de la arteria coronaria, es la mejor opción para pacientes con d-TGA y una válvula pulmonar normal, y se realiza en las primeras semanas de vida. Una vez disminuye la resistencia vascular pulmonar, el ventrículo izquierdo ya no está condicionado a bombear contra la mayor resistencia asociada a la circulación sistémica. TGA-CIA se debe reparar en el periodo neonatal si se identifica a tiempo. Sin embargo, se ha informado de una reparación tardía con éxito (1-2 meses de edad), aunque con un aumento de morbilidad. Los pacientes con la combinación de TGA, CIV y PS pueden haber sido sometidos a una operación de Rastelli. En esta operación se utiliza un parche de comunicación interventricular en ángulo para dirigir la sangre desde el ventrículo izquierdo (VI) a través de la comunicación interventricular hasta la aorta anterior. Este procedimiento también implica la colocación de un conducto desde el ventrículo derecho (VD) hasta la arteria pulmonar (AP). Aunque esta operación da como resultado la restauración del ventrículo izquierdo como ventrículo sistémico, el tratamiento a largo plazo a menudo implica múltiples reoperaciones para el reemplazo del conducto VD a la AP. Aunque ya no se utiliza, principalmente los adultos mayores (>35-40 años) nacidos con D-TGA se han sometido al procedimiento de cambio auricular, ya sea en forma de operación de Senning o de Mustard.[38] El VD es el ventrículo sistémico después de estos procedimientos.[26] Alrededor del 80% de estos pacientes han tenido un buen resultado varias décadas después de la cirugía. Sin embargo, en la gran mayoría de los pacientes adultos se presentan complicaciones a largo plazo, como arritmias y anomalías de la conducción, obstrucción o regurgitación del deflector venoso sistémico o pulmonar y disfunción sistémica del ventrículo derecho que provoca insuficiencia cardíaca.[14] Los adultos que presentan disfunción del VD después de los procedimientos de switch auricular son candidatos para un trasplante de corazón.[26]
Abarca <1% de todas las cardiopatías congénitas. Consta de una comunicación interventricular (CIV) de salida grande y solo 1 arteria grande, el tronco, que surge desde el corazón y que da lugar a las arterias coronarias, las arterias pulmonares y la aorta. Existen cuatro subtipos, basados en la presencia o ausencia de una arteria pulmonar principal, la ramificación de las arterias pulmonares y la formación de la aorta.[44]
[Figure caption and citation for the preceding image starts]: Subtipos de tronco arterioso con comunicación interventricular (tipo A): Tipo A1: la arteria pulmonar principal está presente y se bifurca en las arterias pulmonares izquierda y derecha. Tipo A2: las arterias pulmonares de rama derecha e izquierda surgen de un tronco común. Tipo A3: Una rama de la arteria pulmonar surge del tronco común y la otra está ausente o surge de un CAP o de la aorta. Tipo A4: Tronco con hipoplasia del arco aórtico, coartación o arco aórtico interrumpido y un CAP grandeCalder L et al. Am Heart J 1976 Jul; 92(1):23-38; usado con permiso [Citation ends].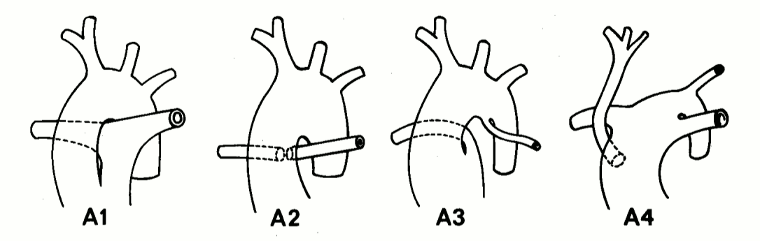
Una microdeleción de 22q11.2 (síndrome de DiGeorge), un cayado aórtico interrumpido y una insuficiencia de la válvula troncal son las anomalías asociadas comunes.
Los pacientes presentan insuficiencia cardíaca congestiva (ICC) o cianosis.
El examen revela un impulso ventricular derecho prominente y un soplo sistólico eyectivo en el borde esternal izquierdo. También puede haber un chasquido de eyección de la válvula troncal. La regurgitación de la válvula troncal tiene como resultado un soplo diastólico decreciente. La interrupción de la aorta y un conducto arterioso permeable (CAP) restrictivo produce una disminución de los pulsos femorales.
En el electrocargiograma (ECG) se observa la dominancia ventricular derecha de la infancia y la radiografía de tórax (RT) revela cardiomegalia con un aumento de las tramas pulmonares. La ecocardiografía transtorácica ayuda a identificar de manera precisa varios aspectos de esta lesión.[3][45][46]
La ICC se puede tratar con diuréticos. La reparación quirúrgica implica el cierre de la CIV, la separación de las circulaciones pulmonar y sistémica, y la colocación de un conducto del ventrículo derecho a la arteria pulmonar. Por lo general, la cirugía se realiza a los 2-3 meses de edad para prevenir el desarrollo de hipertensión pulmonar con resistencia vascular pulmonar elevada. Los pacientes también pueden requerir varias intervenciones con transcatéter o nuevas operaciones para tratar la disfunción del conducto del ventrículo derecho a la arteria pulmonar.[47] La mortalidad quirúrgica es mayor con anomalías asociadas, como la interrupción del cayado aórtico.
Es una malformación poco frecuente de la válvula tricúspide en la que la delaminación anormal de las valvas septal y posterior de la válvula conduce a diversos grados de regurgitación tricúspide, que puede ser grave en una etapa temprana de la vida.[48][49][50] Como consecuencia de la falta de delaminación y dependiendo de su gravedad, una parte del ventrículo derecho (VD) se "atrializa", es decir, se encuentra por debajo del nivel de la válvula tricúspide deformada, y no contribuye al flujo directo. En casos raros, también está presente miopatía ventricular izquierda (VI). En el 80% al 94% de los pacientes, existe una comunicación interauricular concomitante.[50] La cianosis puede producirse debido a la corriente de sangre desoxigenada desde el chorro excéntrico de regurgitación de la válvula tricúspide y a través de la comunicación interauricular.
Los pacientes tienen el primer y segundo ruidos cardíacos con desdoblamiento. La RT revela una cardiomegalia notoria con un borde derecho prominente y un descenso de la vascularidad pulmonar. El electrocardiograma (ECG) muestra ondas "p" picudas en la derivación de extremidades II y un patrón de bloqueo de la rama derecha.[50][51] El síndrome de Wolff-Parkinson-White se ha detectado en al menos el 15% de los pacientes. La ecocardiografía, tanto transtorácica como transesofágica, ayuda a evaluar las anomalías estructurales valvulares y el VD atrializado.[3] La resonancia magnética cardíaca es útil para delinear el grado de atrialización del VD, su tamaño y su función.
Existen varios grados de gravedad anatómica y fisiológica. Los pacientes con cardiomegalia mínima y regurgitación tricúspide pueden llevar una vida normal sin la necesidad de cirugía. Los recién nacidos con formas graves de anomalía de Ebstein requieren prostaglandina E1 para aumentar el flujo sanguíneo pulmonar hasta que se elimine la resistencia vascular pulmonar, y algunos pueden llevar una vida normal sin cirugía. No obstante, el manejo puede resultar difícil en un subconjunto de pacientes con cardiomegalia masiva. La insuficiencia tricuspídea grave es una indicación para cirugía, después de una cuidadosa evaluación hemodinámica.[26]
Las intervenciones quirúrgicas consisten en la reparación, reemplazo o reparación de la válvula tricúspide mediante la operación de cono de Da Silva, que es una cirugía más extensa en la que se repara la válvula tricúspide, se devuelve el anillo a su ubicación anatómica y se reseca el tejido auricular excesivo. Cuando lo realizan operadores expertos, los resultados de la operación del cono en términos de función de la válvula son excelentes.[52]
La CVPAT abarca <1% de todas las cardiopatías congénitas y se produce cuando todas las venas pulmonares se conectan al sistema venoso o a la aurícula izquierda. La CVPAT se puede subdividir en supracardíaca, cardíaca, infracardíaca y mixta, de las cuales la supracardíaca es la más frecuente. La CVPAT provoca que la sangre plenamente oxigenada de los pulmones se dirija de vuelta a la aurícula derecha, y la supervivencia requiere una comunicación interauricular, sin la cual la sangre no puede llegar a la aorta.[53] La CVPAT se puede complicar con la obstrucción de las venas que drenan el confluente de la vena pulmonar, lo que provoca hipertensión pulmonar y congestión.
Los pacientes con venas pulmonares no obstruidas y una gran comunicación intrauricular presentan cianosis leve y aumento del flujo sanguíneo pulmonar. El examen revela un impulso ventricular derecho prominente y un soplo sistólico eyectivo. Una RT muestra cardiomegalia moderada y aumento de las tramas vasculares pulmonares. La forma supracardíaca de la CVPAT revela de manera clásica una configuración de "imagen en ocho". La ecocardiografía puede mostrar una dilatación de la aurícula derecha y del ventrículo derecho, y una confluencia venosa pulmonar posterior.[3]
Los pacientes requieren cirugía pero presentan una excelente supervivencia a largo plazo.
Los pacientes con obstrucción del flujo sanguíneo venoso pulmonar presentan dificultad respiratoria derivada de un edema pulmonar. Es frecuente en recién nacidos que tienen una conexión infracardíaca y los pacientes requieren reparación quirúrgica de emergencia.[53] La mortalidad es de hasta un 40%, a pesar de la cirugía inmediata.
Provoca la ausencia de una comunicación directa entre la aurícula derecha y el ventrículo derecho. Esta anomalía representa aproximadamente el 3% de todas las cardiopatías congénitas (CPC). La supervivencia inicial depende de una comunicación intrauricular. La presentación y el tratamiento dependen de la relación de las arterias grandes (normales o transpuestas), la presencia de una comunicación interventricular (CIV) y la gravedad de la estenosis de la válvula pulmonar.
Normalmente, los pacientes presentan cianosis. El impulso apical puede ser prominente. El segundo ruido cardíaco puede ser único o presentar desdoblamiento. Un soplo sistólico puede deberse a una CIV o estenosis pulmonar, mientras que un soplo apical diastólico medio puede deberse a un aumento del flujo sanguíneo pulmonar e insuficiencia cardíaca congestiva.
La radiografía de tórax (RT) revela cardiomegalia con un borde auricular derecho prominente. Las tramas vasculares pulmonares varían en función del grado de estenosis pulmonar. El electrocardiograma (ECG) se caracteriza por la desviación del eje izquierdo y un asa inicial del plano central girada a la izquierda. Puede haber hipertrofia ventricular izquierda.
Si la fuente del flujo sanguíneo pulmonar no es fiable, el paciente se debe tratar con prostaglandina E1 hasta que se pueda establecer una derivación sistémica-pulmonar. Aquellos pacientes con grandes arterias transpuestas presentan un flujo sanguíneo pulmonar sin restricción y requieren cerclaje de la arteria pulmonar para evitar una enfermedad vascular pulmonar obstructiva y/o edema pulmonar, junto con medicamentos anticongestivos. En última instancia, todos los pacientes se someten a una operación de Fontan en la que el retorno venoso sistémico se conecta directamente a las arterias pulmonares (conexión cavo-pulmonar total).[38] La supervivencia a largo plazo supera el 85% con un seguimiento de 10 años después de la cirugía. Las complicaciones a largo plazo de la operación de Fontan incluyen anomalías en la conducción, arritmia, trastornos de los vasos linfáticos que conducen a bronquitis plástica y enteropatía perdedora de proteínas, hepatopatía congestiva con fibrosis hepática y cirrosis final y fallo de la circulación de Fontan que justifica el trasplante de corazón.
Hace referencia a un espectro de lesiones que incluye tabique ventricular intacto con el subdesarrollo del ventrículo izquierdo y las válvulas mitral y aórtica. Los pacientes presentan frecuentemente coartación aórtica. Los recién nacidos pueden parecer normales durante varios días después del nacimiento, pero dependen de un conducto arterioso permeable (CAP). Una vez se cierra el conducto, desarrollan shock.
El examen revela una disminución en los pulsos de las extremidades inferiores y un aumento del impulso ventricular derecho. La ecocardiografía se utiliza para la evaluación pre y postoperatoria, aunque la visualización de la anatomía de la arteria coronaria puede ser un desafío debido a la hipoplasia aórtica comúnmente asociada.[46]
El SVIH sin tratar es mortal, aunque la mortalidad se ha reducido notablemente en la última década. Los pacientes requieren un abordaje quirúrgico por etapas.[54]
La etapa 1 es un procedimiento de Norwood, que consiste en la reconstrucción del cayado aórtico mediante la arteria pulmonar principal y una derivación Blalock-Taussig modificada que suministra flujo sanguíneo a las ramas de las arterias pulmonares. Recientemente, se ha usado la colocación de una derivación del ventrículo derecho a la arteria pulmonar (Sano) como alternativa a una derivación Blalock-Taussig durante la paliación de la etapa 1.[54] La etapa 2 consiste en la sustitución de la derivación Blalock-Taussig modificada (o derivación Sano) por una anastomosis cavopulmonar bidireccional (Glenn). La etapa 3 consiste en la finalización de la conexión cavopulmonar y la formación de la circulación de Fontan, como se detalla en la sección sobre atresia tricuspídea. En los últimos años, se han utilizado procedimientos híbridos de cateterismo intervencionista/quirúrgico con cerclaje de las arterias pulmonares de las ramas para limitar el flujo sanguíneo pulmonar y el despliegue del stent para mantener la permeabilidad ductal, aunque la utilidad de este abordaje aún no está clara. En ocasiones se realiza un trasplante cardíaco como estrategia alternativa a la cirugía por etapas.[55] Las complicaciones a largo plazo de la circulación de Fontan son similares a las descritas en la sección sobre atresia tricuspídea, y existe cierta evidencia de que en presencia de un ventrículo derecho sistémico pueden producirse en una etapa más temprana.
Incluye 4 subtipos: estenosis de la válvula aórtica, estenosis supravalvular, estenosis subvalvular distinta y estenosis subaórtica de tipo túnel.
La estenosis de la válvula aórtica es la más frecuente y representa el 5% de todas las cardiopatías congénitas (CC).[56] Las valvas o cúspides están generalmente mal formadas o presentan un mayor grosor. Aproximadamente el 2% de la población general tiene una válvula aórtica bicúspide, pero muchas de estas personas nunca desarrollará estenosis de la válvula aórtica o regurgitación valvular aórtica clínicamente significativas. La estenosis supravalvular se produce como una zona de estrechamiento distinto o difuso distal a la unión sinotubular en la aorta ascendente. Se asocia frecuentemente al síndrome de Williams o a un defecto en el gen de la elastina en el cromosoma 7. La estenosis subvalvular distinta se produce cuando hay una cresta fibromuscular que causa obstrucción justo debajo de la válvula aórtica. La turbulencia del flujo sanguíneo puede provocar daños a las valvas de la válvula aórtica, lo que causa una insuficiencia valvular aórtica progresiva. La estenosis subaórtica de tipo túnel hace referencia a un segmento estenótico más largo del tracto de salida cuando se compara con una estenosis subvalvular distinta.
La presentación depende de la gravedad de la obstrucción. La mayoría de los pacientes son asintomáticos. Las pacientes de edad avanzada pueden presentar dolor torácico o síncope. Un subconjunto de pacientes presenta una función ventricular izquierda deficiente, gasto cardíaco bajo y signos de shock e insuficiencia cardíaca congestiva (estenosis aórtica "crítica").
El examen revela un soplo sistólico creciente y decreciente, que se oye mejor en el borde esternal izquierdo con radiación al borde esternal superior derecho. La estenosis moderada o grave tiene como resultado un frémito palpable y pulsos retardados. También se puede oír un chasquido de eyección justo después del primer ruido cardíaco en pacientes con estenosis de la válvula aórtica. El chasquido no varía con la respiración, a diferencia de un chasquido de eyección de la válvula pulmonar. Los pacientes con insuficiencia valvular aórtica concurrente también presentan un soplo diastólico decreciente y una presión de pulso amplia.
El electrocardiograma (ECG) muestra de manera inconsistente evidencia de hipertrofia ventricular izquierda. La estenosis de TSVI grave se asocia al aplanamiento o inversión de las ondas T en las derivaciones V5 y V6.
La radiografía de tórax (RT) puede revelar una aorta prominente como resultado de una dilatación posterior a la estenosis. La ecocardiografía ayuda a evaluar de manera precisa las características morfológicas de la válvula aórtica y calcular el gradiente de presión a lo largo de la TSVI. En la estenosis aórtica valvular, se utiliza el gradiente medio calculado por Doppler para evaluar la gravedad. El gradiente variará con el gasto cardíaco.
Los pacientes con estenosis aórtica "crítica" requieren un alivio de emergencia de la estenosis. Los pacientes sintomáticos también requieren un alivio de la obstrucción, independientemente del grado de la estenosis. Los lactantes con estenosis valvular grave requieren valvuloplastia con globo, aunque puede que sea necesaria la cirugía para una insuficiencia valvular aórtica coexistente. Se considera la valvulotomía quirúrgica o el reemplazo de la válvula en niños mayores y adultos. Los pacientes con estenosis grave, seguidos en el segundo estudio de evolución natural de las cardiopatías congénitas (Second Natural History Study of Congenital Heart Defects), presentaron una tasa de supervivencia a 25 años del 81%.[57] La dilatación de la aorta, ya sea a nivel de la raíz o de la parte ascendente, está presente en el 20% al 30% de los pacientes con válvula aórtica bicúspide, y en pacientes adultos debe reemplazarse cuando alcanza un diámetro de 5.5 cm, o 5 cm si es necesaria una cirugía de la válvula aórtica, o el paciente tiene un aneurisma de aorta torácica hereditario.[58]
Constituye el 5% de las cardiopatías congénitas (CPC) y es más frecuente en hombres; la coartación aórtica se asocia al síndrome de Turner en mujeres. Consiste en un saliente posterior de tejido que sobresale en la aorta y normalmente se describe como yuxtaductal porque se produce a lo largo del conducto arterioso. Por lo general, se asocia con una válvula aórtica bicúspide (entre el 70% y el 75% de los pacientes con coartación aórtica tienen una válvula aórtica bicúspide, pero solo alrededor del 7% de los pacientes con válvula aórtica bicúspide también tienen coartación aórtica) y, con menos frecuencia, con una comunicación interventricular o estenosis aórtica subvalvular.[59]
Puede que los pacientes con obstrucción leve no presenten hasta la adolescencia un soplo cardíaco o hipertensión sistémica. En aquellos pacientes con obstrucción grave, el conducto arterioso permeable (CAP) es la fuente principal del flujo sanguíneo sistémico distal a la obstrucción, lo que significa que los neonatos desarrollan acidosis metabólica y shock cuando se cierra el conducto.
Las extremidades inferiores tienen menor saturación de oxigeno y pulsos decrecientes, además de insuficiencia cardíaca congestiva en pacientes con obstrucción grave. Es importante tomar la presión arterial en ambas extremidades superiores y en 1 pierna antes de realizar el diagnóstico ya que resulta sencillo diagnosticar incorrectamente a pacientes con una coartación proximal a la arteria subclavia izquierda o a aquellos con un origen anómalo de la arteria subclavia derecha. El flujo sanguíneo turbulento por la zona de la coartación produce un soplo sistólico. Los pacientes con una válvula aórtica bicúspide asociada tienen un chasquido de eyección.
Los lactantes, paradójicamente, presentan hipertrofia ventricular derecha en el electrocardiograma (ECG) y la ecocardiografía debido al CAP que suministra a la aorta distal a la obstrucción. Los pacientes de edad avanzada presentan signos de hipertrofia ventricular izquierda. La tomografía computarizada (TC) y la imagen por resonancia magnética (IRM) pueden resultar útiles para definir la anatomía de la coartación en el implante de stent. El método de reparación quirúrgica más utilizado es una anastomosis extendida de extremo a extremo, que cuando es realizada por operadores expertos, tiene una excelente duración y resultados a largo plazo.[60] Otras técnicas quirúrgicas incluyen la reparación del colgajo subclavio y el uso de un injerto protésico. La reparación en el período neonatal se asocia con una mayor incidencia de recurrencia, que se trata mejor mediante la colocación de un stent. Otras complicaciones a largo plazo incluyen la formación de aneurismas en el sitio de la anastomosis y la disección aórtica. A pesar de su naturaleza localizada, es bien sabido que la coartación aórtica causa disfunción endotelial sistémica, y muchos pacientes corren el riesgo de desarrollar hipertensión de inicio temprano, lo que puede conducir a eventos adversos cardiovasculares como accidente cerebrovascular, enfermedad arterial periférica y cardiopatía isquémica.[61] Hasta un 10% de los pacientes desarrollan microaneurismas vasculares intracraneales (a veces denominados "aneurismas en baya"), especialmente en la zona anatómica del círculo de Willis.[62] El seguimiento de por vida, incluso en pacientes que no tienen una obstrucción residual, es extremadamente importante.
Para obtener más información, consulte Coartación aórtica.
Representa hasta el 8% de todas las cardiopatías congénitas y se detecta comúnmente en pacientes con el síndrome de Noonan. La mayoría de los niños presentan un soplo asintomático. No obstante, los neonatos con estenosis crítica de la válvula pulmonar presentan una cianosis derivada de una derivación de derecha a izquierda de la sangre en el nivel auricular.[63]
El examen revela un aumento del impulso ventricular derecho, un chasquido que sigue al primer ruido cardíaco y que varía con la respiración, un segundo ruido cardíaco con desdoblamiento de normal a amplio que depende de la gravedad y un soplo eyectivo creciente y decreciente. Con el aumento de la gravedad de la estenosis, el chasquido de eyección pulmonar se produce de manera más temprana en la sístole; en los casos más graves, el chasquido puede surgir con el primer ruido cardíaco y volverse inaudible.[49] Por el contrario, el segundo ruido cardíaco se desdobla de manera más amplia con el aumento de la gravedad de la estenosis y puede volverse fijo en estenosis grave. En una estenosis muy grave, el componente pulmonar del segundo ruido puede volverse difícil de oír debido al fuerte soplo que desemboca en la diástole. Se puede oír un cuarto ruido cardíaco en pacientes con insuficiencia ventricular derecha.[49]
El electrocardiograma (ECG) puede revelar una desviación del eje derecho y una radiografía de tórax (RT) puede mostrar signos de agrandamiento ventricular derecho. Normalmente, no hay dilatación posterior a la estenosis de las arterias pulmonares.
El tratamiento de elección es la valvuloplastia con globo y, a los 10 años de seguimiento, el 84% de los pacientes no requieren más intervenciones.[63] El resultado a largo plazo es excelente. Si finalmente se necesita un reemplazo valvular, se puede realizar quirúrgicamente o mediante un abordaje transcatéter.
Para más información, consulte Estenosis pulmonar.
Las personas con CC pueden correr un mayor riesgo de sufrir una infección más grave por COVID-19, en particular aquellas con características anatómicas y fisiológicas más graves de CC.[64][65] Los defectos biventriculares simples, los defectos ventriculares complejos y la cirugía cardíaca se asocian con COVID-19 grave.[66] Se han publicado recomendaciones para la prevención y el manejo de la COVID-19 en adultos con CPC, basadas en la estratificación del riesgo.[65][67]
Se recomienda continuar con los medicamentos cardíacos, como la aspirina, los inhibidores de la enzima convertidora de angiotensina (ECA), los antagonistas de los receptores de angiotensina II, los betabloqueantes, los diuréticos y los medicamentos antiarrítmicos durante la enfermedad por COVID-19, a menos que exista una contraindicación clara.[64]
Para obtener más información, consulte el apartado Enfermedad por coronavirus 2019 (COVID-19).
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad