Etiología
La pancitopenia puede deberse a una disminución en la producción celular de la médula ósea o a un fallo medular, trastornos clonales de la hematopoyesis, un aumento en la destrucción o el secuestro no inmunomediado, o bien a una destrucción inmunomediada de células sanguíneas.[Figure caption and citation for the preceding image starts]: Tabla de etiologías de pancitopenia (LES: lupus eritematoso sistémico, CMV: citomegalovirus, VEB: virus Epstein-Barr, EICH: enfermedad de injerto contra huésped)De la colección de Jeff K. Davies [Citation ends].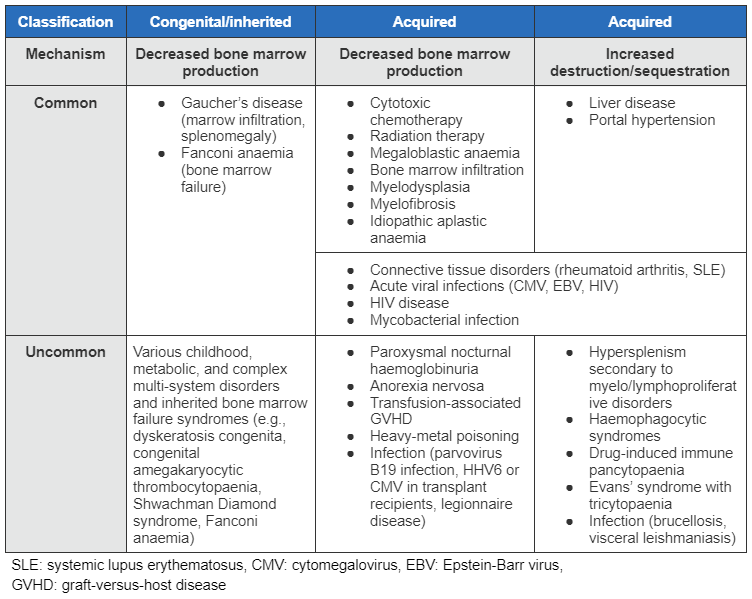
Disminución en la producción de la médula ósea
La médula ósea es el sitio de producción de eritrocitos, leucocitos y megacariocitos, de los cuales se forman las plaquetas. Una vez generadas las células, estas se liberan a la circulación periférica. Este proceso requiere una actividad adecuada de las células madre hematopoyéticas y un ambiente funcional del estroma de la médula ósea. La alta tasa proliferativa de la médula requiere un estado nutricional adecuado, en particular de vitamina B12 y ácido fólico, así como cantidades mínimas de otros elementos.
Quimioterapia
La causa más frecuente de pancitopenia transitoria en todos los grupos de edad es la quimioterapia citotóxica y la radioterapia.
Es infrecuente que la pancitopenia relacionada con la quimioterapia presente un dilema diagnóstico y suele resolverse en 1 o 2 semanas (a menos que exista un síndrome de fallo medular hereditario no conocido). Algunos individuos pueden presentar defectos en la proliferación conocidos o desconocidos o una farmacogenética particular, que pueden predisponerles a una pancitopenia más grave y de mayor duración. Algunos regímenes están asociados a períodos de pancitopenia significativamente más prolongados. Debe investigarse la pancitopenia postquimioterapia que sea más prolongada de lo esperado.
Anemia megaloblástica
Aunque la mayoría de los casos de anemia megaloblástica causan una anemia macrocítica sin leucopenia ni trombocitopenia, la anemia megaloblástica grave puede derivar en una pancitopenia. La anemia megaloblástica con frecuencia se debe a un déficit de vitamina B12 (p. ej., anemia perniciosa, una afección autoinmune en la que los autoanticuerpos interfieren en la función del factor intrínseco, que se requiere para la absorción de la vitamina B12 dentro del tracto gastrointestinal). Con menor frecuencia, la deficiencia de vitamina B12 se produce debido a un déficit de la dieta (en personas veganas) o por una malabsorción en el intestino.
La deficiencia de ácido fólico, casi siempre dietaria en origen, también produce una anemia megaloblástica.
Infiltración de médula ósea
La infiltración de la médula ósea es una causa frecuente de pancitopenia y generalmente se debe a una enfermedad maligna. En general, el infiltrado es celular y puede ser de origen hematológico (p. ej., leucemia mieloide aguda y leucemia linfoblástica, mieloma, linfoma no Hodgkin, tricoleucemia, leucemia linfocítica crónica y mielofibrosis) o tener un origen maligno no-hematológico (p. ej., mama, pulmón, riñón, próstata y tiroides).
En niños, la pancitopenia puede deberse a neuroblastoma, rabdomiosarcoma, sarcoma de Ewing y retinoblastoma.[1]
Enfermedades de depósito lisosomal
Las enfermedades de depósito lisosomal (p. ej., enfermedad de Gaucher) pueden infiltrar la médula, lo que causa pancitopenia. El infiltrado puede ser en su mayoría fibrosis de reticulina, que también está asociada a enfermedades malignas.
Los pacientes con enfermedad de Gaucher pueden presentar esplenomegalia masiva e hiperesplenismo funcional, además de infiltración de la médula ósea.[2]
Otras causas
Entre las causas menos frecuentes de pancitopenia que se deben a una disminución en la producción de células sanguíneas en la médula ósea, se incluyen la anorexia nerviosa, la enfermedad de injerto contra huésped asociada a la transfusión en pacientes inmunosuprimidos y la intoxicación por metales pesados (p. ej., arsénico).[3]
Infecciones, como la infección por VIH, se han relacionado con la pancitopenia secundaria a una subproducción (que en la mayoría de los casos afecta solo a la producción de eritrocitos), como sucede con el parvovirus en personas con condiciones predisponentes específicas (anemia hemolítica; predominando la anemia falciforme y la esferocitosis hereditaria).[4]
Trastornos clonales de hematopoyesis
El síndrome mielodisplásico (SMD) es un trastorno clonal adquirido frecuente de las células hematopoyéticas, que se caracteriza por una hematopoyesis displásica e ineficaz y una propensión a evolucionar a leucemia mieloide aguda. Las personas adultas suelen estar en la década de los 70 años en el momento del diagnóstico.[5][6] En los niños, se suele utilizar el término citopenias refractarias de la infancia (CRI o RCC, por sus siglas en inglés). Los niños con SMD/CRI suelen tener una predisposición germinal subyacente (IBMFS, RUNX1, ANKRD26, ETV6, GATA2 y SAMD9/SAMDL9) a padecer SMD en aproximadamente el 30% al 40% de los casos.[7]
La médula ósea puede ser hipercelular o hipocelular. En ambos casos, existe frecuentemente una pancitopenia en sangre periférica. Además de la producción reducida o inadecuada de células sanguíneas en la médula, a veces existe un mecanismo inmunomediado que contribuye a la pancitopenia en sangre periférica en el SMD.
La hemoglobinuria paroxística nocturna (HPN) es un trastorno clonal adquirido poco frecuente (tasa de incidencia de 5,7 por 1.000.000 personas-año) de las células hematopoyéticas, causado por una mutación somática del gen del fosfatidilinositol glicano del grupo A situado en el cromosoma X y que da lugar a una expresión deficiente de las proteínas ancladas al glicosilfosfatidilinositol.[8][9] La HPN se caracteriza clínicamente por la hemólisis intravascular y la trombosis y es frecuente la evolución a pancitopenia (probablemente surja de una combinación de la producción reducida de la médula ósea, a consecuencia de defectos adquiridos en las células madre hematopoyéticas y a la destrucción celular). Existe un solapamiento en las características clínicas y de laboratorio entre los pacientes con HPN y los que padecen anemia aplásica idiopática (AAI) e incluso SMD.[Figure caption and citation for the preceding image starts]: Síndrome mielodisplásico hipoplásico con normoblastos displásicosMorris Edelman, MD and Peihong Hsu, MD [Citation ends].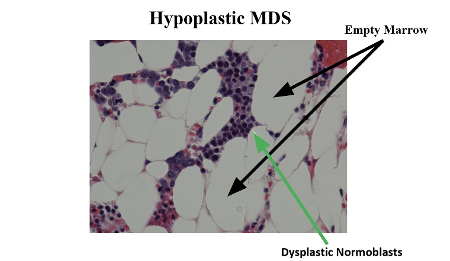
Insuficiencia medular
Los síndromes de insuficiencia medular hereditaria (IBMFS) y congénita se presentan con mayor frecuencia en la niñez, aunque el diagnóstico en la adultez está aumentando a consecuencia de la creación de conciencia y la mayor cantidad de pruebas.
Anemia de Fanconi: es principalmente un trastorno autosómico recesivo (se ha descrito ocasionalmente una herencia dominante y ligada al cromosoma X) en el que unas 20 proteínas disfuncionales causan una disminución en la hematopoyesis y un fallo medular.[10] La anemia de Fanconi se caracteriza de forma variable por estatura baja, hiperpigmentación, alteraciones esqueléticas, mayor incidencia de tumores sólidos y leucemia y un aumento en la sensibilidad celular a agentes que dañan el ADN.[11][12][13]
Disqueratosis congénita (DC): se hereda como trastorno ligado al cromosoma X, autosómico dominante o autosómico recesivo, se debe a lesiones genéticas que comprometen la integridad de los telómeros, con una pérdida de la autorrenovación y la regeneración celular.[14] Las mutaciones en los 14 genes asociadas a la biología de los telómeros se pueden identificar en la mayoría de los pacientes con características clínicas de DC clásica.[15][16][17] La DC clásica se define por distrofia de las uñas, leucoplasia de la mucosa y cambios en la pigmentación de la piel, cuya gravedad puede ser desde prácticamente inexistente a grave.[13] Entre las otras alteraciones se incluyen insuficiencia medular, calvicie y cabello canoso prematuros, estenosis uretral, producción excesiva de lágrimas y fibrosis pulmonar.[18]
Anemia aplásica idiopática (adquirida) (AAI): es una afección adquirida poco frecuente (2-6 casos por millón de habitantes en la población general). El diagnóstico de AAI requiere la presencia de pancitopenia en combinación con una disminución en la celularidad de la médula ósea sin infiltración ni fibrosis.[19] Por lo tanto, la AAI es un diagnóstico de exclusión y se debe diferenciar cuidadosamente del SMD hipocelular y de los síndromes de fallo medular congénitos y hereditarios.[20] Algunos pacientes tienen antecedentes de infección viral, hepatitis o exposición a fármacos. La AAI grave (donde la neutropenia y la trombocitopenia son más profundas) es una afección potencialmente mortal.
Otras citopenias hereditarias infrecuentes de una única línea celular: la anemia de Diamond Blackfan (DBA), el síndrome de Shwachman Diamond (SDS) y la trombocitopenia amegacariocítica (TA) pueden evolucionar a pancitopenia.[13]
Mediante el uso de la secuenciación del exoma completo y del genoma completo, se han identificado mutaciones genéticas poco frecuentes en el fallo medular hereditario en pacientes con fallo medular con sospecha de origen hereditario, pero con un diagnóstico no aclarado.[21][Figure caption and citation for the preceding image starts]: Anemia aplásica: la médula ósea normocelular se muestra a la izquierda; y la médula vacía, típica de la anemia aplásica congénita o adquirida, a la derecha.Morris Edelman, MD and Peihong Hsu, MD [Citation ends].
Aumento en la destrucción o el secuestro
La mayoría de los casos de pancitopenia que están acompañados de una producción adecuada de células sanguíneas en la médula ósea derivan de un aumento en el secuestro de células sanguíneas dentro del bazo. Entre las enfermedades que causan pancitopenia a partir de un hiperesplenismo funcional se incluyen:
Hepatopatía (con hipertensión portal asociada) causada por cirrosis hepática alcohólica, hepatitis B y C crónica, hepatitis autoinmunitaria o hipertensión portal idiopática.
Enfermedades mieloproliferativas (p. ej., la leucemia mieloide crónica se puede presentar con una esplenomegalia masiva que produce pancitopenia a pesar de la producción adecuada de células sanguíneas en la médula ósea). Estas enfermedades rara vez aparecen en niños.
Infecciones agudas y crónicas que producen hiperesplenismo (p. ej., brucelosis y leishmaniasis visceral). Es particularmente relevante considerar el historial de viajes y de exposición.
Síndromes hemofagocíticos, un grupo heterogéneo de trastornos caracterizados por un aumento en la actividad de macrófagos o histiocitos en la médula ósea y en otros órganos. La hepatomegalia y la esplenomegalia son características clínicas frecuentes. Los síndromes hemofagocíticos pueden clasificarse como primarios (en los que el síndrome hemofagocítico domina las características clínicas de la afección, como en la linfohistiocitosis hemofagocítica primaria [pHLH]), que suelen ser de origen genético, o pueden ser reactivos a afecciones sistémicas con una serie de otras características clínicas (p. ej., trastornos autoinmunitarios, linfoma de células T, denominado síndrome de activación de macrófagos). Estas diferencias pueden ser difíciles de distinguir en algunos casos, pero las pruebas genéticas están disponibles para una serie de genes implicados en la pHLH.[22]
Destrucción inmunomediada de células sanguíneas
La pancitopenia inmunitaria inducida por fármacos se produce cuando se generan anticuerpos con reactividad cruzada para fármacos y células hematopoyéticas. Se asocia con mayor frecuencia a la quinina, las sulfamidas, el metotrexato y la rifampicina.[23]
La pancitopenia inmune se puede observar en hasta el 20% de los pacientes con síndrome de Evans (la combinación clásica de trombocitopenia autoinmune y anemia hemolítica), que se ve con mayor frecuencia en niños que en adultos.[24] Un número significativo de personas con síndrome de Evans tienen síndrome linfoproliferativo autoinmune subyacente.
El síndrome linfoproliferativo autoinmune (SLPA) es un trastorno hereditario que se produce debido a mutaciones que inhiben la apoptosis en la regulación de la respuesta inmune. Se han notificado casos leves, lo que sugiere que la incidencia está significativamente infraestimada. El síndrome linfoproliferativo autoinmune (SLPA) se caracteriza por una linfoproliferación generalmente benigna (linfadenopatía y esplenomegalia) y autoinmunidad, dirigida con mayor frecuencia a las células del linaje mieloide (eritrocitos, granulocitos y plaquetas), aunque con menor frecuencia hay otros órganos involucrados (p. ej., hepatitis autoinmune).[25]
Pancitopenia causada por combinación
Muchas enfermedades relacionadas con la pancitopenia se deben a una combinación de producción reducida de la médula ósea y a una destrucción o secuestro aumentado de células sanguíneas. Entre ellas se incluyen:
Trastornos del tejido conjuntivo (con mayor frecuencia artritis reumatoide y lupus eritematoso sistémico)
Infección aguda por citomegalovirus
Infección por micobacterias
Mononucleosis infecciosa
VIH
Síndrome de Felty (artritis reumatoide, esplenomegalia y neutropenia).
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad