Initial clinical manifestations are variable, ranging from lack of symptoms to complications of end-stage liver disease. An increasing proportion are identified through workup of asymptomatic elevated liver enzymes detected on routine laboratory testing.[5]Kaplan GG, Laupland KB, Butzner D, et al. The burden of large and small duct primary sclerosing cholangitis in adults and children: a population-based analysis. Am J Gastroenterol. 2007 May;102(5):1042-9.
http://www.ncbi.nlm.nih.gov/pubmed/17313496?tool=bestpractice.com
[6]Bambha K, Kim WR, Talwalkar J, et al. Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a United States community. Gastroenterology. 2003 Nov;125(5):1364-9.
http://www.ncbi.nlm.nih.gov/pubmed/14598252?tool=bestpractice.com
Diagnosis is based on a combination of clinical features, cholestatic liver tests, cholangiographic findings, and histologic findings. However, it is often delayed from the time of presentation due to subtle symptoms and an insidious course.
Diagnosis requires exclusion of secondary sclerosing cholangitis (other conditions that may cause bile duct abnormalities and similar clinical and cholangiographic syndromes, e.g., surgical trauma to the bile duct, choledocholithiasis, recurrent pancreatitis, caustic injury from intra-arterial chemotherapy, or malignancy).[25]Abdalian R, Heathcote EJ. Sclerosing cholangitis: a focus on secondary causes. Hepatology. 2006 Nov;44(5):1063-74.
http://www.ncbi.nlm.nih.gov/pubmed/17058222?tool=bestpractice.com
Sudden and marked deterioration in clinical status or biochemical tests may indicate the presence of complications such as dominant/relevant stricture or cholangiocarcinoma.
Patients with advanced liver disease or cirrhosis should be referred to a liver transplantation center for evaluation and management.
Historical factors
About half of patients are asymptomatic.[4]Kingham JG, Kochar N, Gravenor MB. Incidence, clinical patterns and outcomes of primary sclerosing cholangitis in South Wales, United Kingdom. Gastroenterology 2004 Jun;126(7):1929-30.
http://www.ncbi.nlm.nih.gov/pubmed/15188211?tool=bestpractice.com
Common presenting symptoms include fatigue, upper abdominal pain, and pruritus. Fatigue is very nonspecific but may be present at the time of diagnosis.[5]Kaplan GG, Laupland KB, Butzner D, et al. The burden of large and small duct primary sclerosing cholangitis in adults and children: a population-based analysis. Am J Gastroenterol. 2007 May;102(5):1042-9.
http://www.ncbi.nlm.nih.gov/pubmed/17313496?tool=bestpractice.com
[11]Bergquist A, Said K, Broome U. Changes over a 20-year period in the clinical presentation of primary sclerosing cholangitis in Sweden. Scand J Gastroenterol. 2007 Jan;42(1):88-93.
http://www.ncbi.nlm.nih.gov/pubmed/17190768?tool=bestpractice.com
Abdominal pain is usually nonspecific and located in the right upper quadrant or epigastrium.[5]Kaplan GG, Laupland KB, Butzner D, et al. The burden of large and small duct primary sclerosing cholangitis in adults and children: a population-based analysis. Am J Gastroenterol. 2007 May;102(5):1042-9.
http://www.ncbi.nlm.nih.gov/pubmed/17313496?tool=bestpractice.com
[8]Tischendorf JJ, Hecker H, Kruger M, et al. Characterization, outcome, and prognosis in 273 patients with primary sclerosing cholangitis: a single center study. Am J Gastroenterol. 2007 Jan;102(1):107-14.
http://www.ncbi.nlm.nih.gov/pubmed/17037993?tool=bestpractice.com
[11]Bergquist A, Said K, Broome U. Changes over a 20-year period in the clinical presentation of primary sclerosing cholangitis in Sweden. Scand J Gastroenterol. 2007 Jan;42(1):88-93.
http://www.ncbi.nlm.nih.gov/pubmed/17190768?tool=bestpractice.com
Pruritus is usually generalized and intermittent, although it can also be severe and interfere with sleep and daily activities. Patients with liver disease may also complain of yellowing skin; pale, offensive-smelling stools that are difficult to flush; blood in vomit; confusion; and an increased abdominal girth.
Past medical history may reveal inflammatory bowel disease (typically ulcerative colitis but also Crohn disease), with estimates indicating its presence in 50% to more than 80% of patients - it is estimated that 0.6% to 4.3% of patients with inflammatory bowel disease have PSC.[2]European Association for the Study of the Liver. EASL clinical practice guidelines on sclerosing cholangitis. J Hepatol. 2022 Sep;77(3):761-806.
https://www.doi.org/10.1016/j.jhep.2022.05.011
http://www.ncbi.nlm.nih.gov/pubmed/35738507?tool=bestpractice.com
[3]Bowlus CL, Arrivé L, Bergquist A, et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2023 Feb 1;77(2):659-702.
https://www.doi.org/10.1002/hep.32771
http://www.ncbi.nlm.nih.gov/pubmed/36083140?tool=bestpractice.com
[9]Tabibian JH, Ali AH, Lindor KD. Primary sclerosing cholangitis, part 1: epidemiology, etiopathogenesis, clinical features, and treatment. Gastroenterol Hepatol (N Y). 2018 May;14(5):293-304.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6034608
http://www.ncbi.nlm.nih.gov/pubmed/29991937?tool=bestpractice.com
[13]Karlsen TH, Folseraas T, Thorburn D, et al. Primary sclerosing cholangitis - a comprehensive review. J Hepatol. 2017 Aug 10;67(6):1298-323.
https://www.journal-of-hepatology.eu/article/S0168-8278(17)32196-7/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/28802875?tool=bestpractice.com
Inflammatory bowel disease may develop before or after PSC, and is frequently characterized by pancolitis with rectal sparing and backwash ileitis.[6]Bambha K, Kim WR, Talwalkar J, et al. Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a United States community. Gastroenterology. 2003 Nov;125(5):1364-9.
http://www.ncbi.nlm.nih.gov/pubmed/14598252?tool=bestpractice.com
[11]Bergquist A, Said K, Broome U. Changes over a 20-year period in the clinical presentation of primary sclerosing cholangitis in Sweden. Scand J Gastroenterol. 2007 Jan;42(1):88-93.
http://www.ncbi.nlm.nih.gov/pubmed/17190768?tool=bestpractice.com
[24]Loftus EV Jr, Harewood GC, Loftus CG, et al. PSC-IBD: a unique form of inflammatory bowel disease associated with primary sclerosing cholangitis. Gut. 2005 Jan;54(1):91-6.
https://gut.bmj.com/content/54/1/91.long
http://www.ncbi.nlm.nih.gov/pubmed/15591511?tool=bestpractice.com
Physical examination
Presenting signs may include excoriations (from pruritus), weight loss (from fat malabsorption, active inflammatory bowel disease, and/or advanced liver disease), jaundice, splenomegaly, ascites, encephalopathy, esophageal variceal bleeding, and/or fever (from episodic bacterial cholangitis).
Routine laboratory testing
Although laboratory tests are not diagnostic of PSC, abnormalities (particularly elevated serum alkaline phosphatase) may suggest or support the diagnosis.
Liver function tests
Required for all patients presenting with the above symptoms and in patients with a history of inflammatory bowel disease, even if they are asymptomatic.
About 75% of patients demonstrate a cholestatic pattern.[3]Bowlus CL, Arrivé L, Bergquist A, et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2023 Feb 1;77(2):659-702.
https://www.doi.org/10.1002/hep.32771
http://www.ncbi.nlm.nih.gov/pubmed/36083140?tool=bestpractice.com
Serum alkaline phosphatase is derived from liver and bone and is elevated in the majority of patients with PSC, although it can be normal in up to 8.5% of patients, and is more often normal in children.[26]Broomé U, Olsson R, Lööf L, et al. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut. 1996 Apr;38(4):610-5.
https://gut.bmj.com/content/gutjnl/38/4/610.full.pdf
http://www.ncbi.nlm.nih.gov/pubmed/8707097?tool=bestpractice.com
[27]Wilschanski M, Chait P, Wade JA, et al. Primary sclerosing cholangitis in 32 children: clinical, laboratory, and radiographic features, with survival analysis. Hepatology. 1995 Nov;22(5):1415-22.
http://www.ncbi.nlm.nih.gov/pubmed/7590657?tool=bestpractice.com
Elevated alkaline phosphatase is accompanied by elevated serum gamma-GT level in liver disorders. An elevated serum alkaline phosphatase with a normal gamma-GT should prompt evaluation for bone diseases.
Most patients have mild to moderate elevation in serum aminotransferase levels (typically 2-3 times the upper limit of normal). Predominant aminotransferase levels, or levels more than 5 times the upper limit of normal, may suggest overlapping autoimmune hepatitis, but precise criteria for PSC-autoimmune hepatitis overlap are not available.[3]Bowlus CL, Arrivé L, Bergquist A, et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2023 Feb 1;77(2):659-702.
https://www.doi.org/10.1002/hep.32771
http://www.ncbi.nlm.nih.gov/pubmed/36083140?tool=bestpractice.com
Bilirubin levels fluctuate in the early stages of disease and are more commonly persistently elevated in advanced disease.
Albumin and prothrombin times are usually within normal ranges because the majority of patients have intact liver synthetic function at the time of diagnosis (although albumin may be low due to concomitant active inflammatory bowel disease, and low in advanced liver disease).
Thrombocytopenia (platelet count <150,000 mm³) on CBC indicates advanced liver disease.[28]Afdhal N, McHutchison J, Brown R, et al. Thrombocytopenia associated with chronic liver disease. J Hepatol. 2008 Jun;48(6):1000-7.
https://www.journal-of-hepatology.eu/article/S0168-8278(08)00221-3/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/18433919?tool=bestpractice.com
Additional laboratory testing
Autoantibodies
There are no autoantibodies specific to (or diagnostic of) PSC. Serum autoantibodies should not, therefore, be routinely tested for in patients with possible PSC for diagnostic or risk-stratification purposes.[2]European Association for the Study of the Liver. EASL clinical practice guidelines on sclerosing cholangitis. J Hepatol. 2022 Sep;77(3):761-806.
https://www.doi.org/10.1016/j.jhep.2022.05.011
http://www.ncbi.nlm.nih.gov/pubmed/35738507?tool=bestpractice.com
Autoantibody testing may be appropriate when the following are considerations:
primary biliary cholangitis (antimitochondrial antibody is nearly always present in patients with primary biliary cholangitis, and is notably absent in patients with PSC)
PSC-autoimmune hepatitis overlap syndrome (antinuclear antibody [ANA] and anti-smooth muscle antibody).
The clinical significance of autoantibodies that may be present in patients with PSC (e.g., atypical antineutrophil cytoplasmic autoantibody (ANCA), ANA, anti-smooth muscle antibody, rheumatoid factor) is unclear.
ANA and anti-smooth muscle antibody are present in 8% to 77% and 0% to 83%, respectively, of patients with PSC.[16]Hov JR, Boberg KM, Karlsen TH. Autoantibodies in primary sclerosing cholangitis. World J Gastroenterol. 2008 Jun 28;14(24):3781-91.
http://www.ncbi.nlm.nih.gov/pubmed/18609700?tool=bestpractice.com
Presence of serum autoantibodies should not exclude a diagnosis of PSC (or suggest an alternative diagnosis) in a patient whose clinical picture is otherwise consistent with PSC.
Immunoglobulins
Usually performed in patients with a clinical history and abnormal liver tests suggestive of autoimmune hepatitis or primary biliary cholangitis.
Not an essential part of laboratory evaluation for PSC, but if IgG or IgM is elevated and clinical suspicion is high, PSC should not be ruled out.
Elevated IgG is, however, more commonly associated with autoimmune hepatitis and elevated IgM is more commonly associated with primary biliary cholangitis.[29]Boberg KM, Aadland E, Jahnsen J, et al. Incidence and prevalence of primary biliary cirrhosis, primary sclerosing cholangitis, and autoimmune hepatitis in a Norwegian population. Scand J Gastroenterol. 1998 Jan;33(1):99-103.
http://www.ncbi.nlm.nih.gov/pubmed/9489916?tool=bestpractice.com
Elevated levels of IgG4 have been associated with a multiorgan lymphoplasmocytic sclerosing disease that can include a sclerosing cholangitis. IgG4-associated cholangitis is often associated with autoimmune pancreatitis and responds readily to corticosteroid immunosuppression. Up to 15% of patients with PSC have elevated IgG4 serum levels.[2]European Association for the Study of the Liver. EASL clinical practice guidelines on sclerosing cholangitis. J Hepatol. 2022 Sep;77(3):761-806.
https://www.doi.org/10.1016/j.jhep.2022.05.011
http://www.ncbi.nlm.nih.gov/pubmed/35738507?tool=bestpractice.com
[3]Bowlus CL, Arrivé L, Bergquist A, et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2023 Feb 1;77(2):659-702.
https://www.doi.org/10.1002/hep.32771
http://www.ncbi.nlm.nih.gov/pubmed/36083140?tool=bestpractice.com
A diagnosis of IgG4-associated sclerosing cholangitis is suggested by an IgG4 >5.6 g/L, whereas it can be excluded by a serum IgG4 of 1.4 to 2.8 g/L in combination with a IgG4/IgG1 ratio of <0.24.[2]European Association for the Study of the Liver. EASL clinical practice guidelines on sclerosing cholangitis. J Hepatol. 2022 Sep;77(3):761-806.
https://www.doi.org/10.1016/j.jhep.2022.05.011
http://www.ncbi.nlm.nih.gov/pubmed/35738507?tool=bestpractice.com
[3]Bowlus CL, Arrivé L, Bergquist A, et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2023 Feb 1;77(2):659-702.
https://www.doi.org/10.1002/hep.32771
http://www.ncbi.nlm.nih.gov/pubmed/36083140?tool=bestpractice.com
US guidance recommends testing all patients with possible PSC for IgG4, whereas European guidelines recommend determining serum IgG4 levels in all adults with large-duct PSC at the time of diagnosis.[2]European Association for the Study of the Liver. EASL clinical practice guidelines on sclerosing cholangitis. J Hepatol. 2022 Sep;77(3):761-806.
https://www.doi.org/10.1016/j.jhep.2022.05.011
http://www.ncbi.nlm.nih.gov/pubmed/35738507?tool=bestpractice.com
[3]Bowlus CL, Arrivé L, Bergquist A, et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2023 Feb 1;77(2):659-702.
https://www.doi.org/10.1002/hep.32771
http://www.ncbi.nlm.nih.gov/pubmed/36083140?tool=bestpractice.com
[30]Chapman R, Fevery J, Kalloo A, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010 Feb;51(2):660-78.
https://aasldpubs.onlinelibrary.wiley.com/doi/full/10.1002/hep.23294
http://www.ncbi.nlm.nih.gov/pubmed/20101749?tool=bestpractice.com
Copper
Urine copper levels are usually measured in patients with a clinical history and abnormal liver tests suggestive of Wilson disease.
Not a routine part of laboratory evaluation of PSC, but as elevated levels can be seen in patients with any form of cholestatic liver disease, PSC should not be excluded if levels are elevated.
Ceruloplasmin
If urine copper is elevated, ceruloplasmin level may be helpful in differentiating PSC and Wilson disease.
Ceruloplasmin has been reported to be high in patients with PSC, but is typically low in patients with Wilson disease.
Imaging
Imaging of the biliary tree is essential for establishing diagnosis.
Abdominal ultrasound
Noninvasive initial test to investigate for abnormal (dilated) bile ducts and stones, and may also provide evidence of more advanced liver disease.[2]European Association for the Study of the Liver. EASL clinical practice guidelines on sclerosing cholangitis. J Hepatol. 2022 Sep;77(3):761-806.
https://www.doi.org/10.1016/j.jhep.2022.05.011
http://www.ncbi.nlm.nih.gov/pubmed/35738507?tool=bestpractice.com
Does not provide adequate evaluation of the biliary tree for diagnosis or exclusion of PSC.
Computed tomography (CT)
Abdominal CT scanning may reveal bile duct thickening, focal or saccular intrahepatic duct dilation, and lymphadenopathy. Mass lesions suggestive of cholangiocarcinoma may also be identified.[31]Walker SL, McCormick PA. Diagnosing cholangiocarcinoma in primary sclerosing cholangitis: an "evidence based radiology" review. Abdom Imaging. 2008 Jan-Feb;33(1):14-7.
http://www.ncbi.nlm.nih.gov/pubmed/17874262?tool=bestpractice.com
[32]Campbell WL, Peterson MS, Federle MP, et al. Using CT and cholangiography to diagnose biliary tract carcinoma complicating primary sclerosing cholangitis. AJR Am J Roentgenol. 2001 Nov;177(5):1095-100.
https://www.ajronline.org/doi/full/10.2214/ajr.177.5.1771095
http://www.ncbi.nlm.nih.gov/pubmed/11641179?tool=bestpractice.com
[33]Ament AE, Haaga JR, Wiedenmann SD, et al. Primary sclerosing cholangitis: CT findings. J Comput Assist Tomogr. 1983 Oct;7(5):795-800.
http://www.ncbi.nlm.nih.gov/pubmed/6886129?tool=bestpractice.com
CT scans are useful in patients with recurrent symptoms or laboratory findings indicating worsening disease suggestive of possible cholangiocarcinoma.
Cholangiography
Both magnetic resonance cholangiopancreatography (MRCP) and endoscopic retrograde cholangiopancreatography (ERCP) can establish a diagnosis and provide information on the distribution and extent of disease.
MRCP is the preferred diagnostic test for suspected PSC in adults and children.[2]European Association for the Study of the Liver. EASL clinical practice guidelines on sclerosing cholangitis. J Hepatol. 2022 Sep;77(3):761-806.
https://www.doi.org/10.1016/j.jhep.2022.05.011
http://www.ncbi.nlm.nih.gov/pubmed/35738507?tool=bestpractice.com
[3]Bowlus CL, Arrivé L, Bergquist A, et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2023 Feb 1;77(2):659-702.
https://www.doi.org/10.1002/hep.32771
http://www.ncbi.nlm.nih.gov/pubmed/36083140?tool=bestpractice.com
[34]Lindor KD, Kowdley KV, Harrison ME; American College of Gastroenterology. ACG clinical guideline: primary sclerosing cholangitis. Am J Gastroenterol. 2015 May;110(5):646-59.
https://gi.org/guideline/primary-sclerosing-cholangitis
http://www.ncbi.nlm.nih.gov/pubmed/25869391?tool=bestpractice.com
MRCP is noninvasive; has comparable performance to ERCP, with high sensitivity and specificity for diagnosis; and is not associated with risk of pancreatitis, bleeding, cholangitis, or perforation.[30]Chapman R, Fevery J, Kalloo A, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010 Feb;51(2):660-78.
https://aasldpubs.onlinelibrary.wiley.com/doi/full/10.1002/hep.23294
http://www.ncbi.nlm.nih.gov/pubmed/20101749?tool=bestpractice.com
[35]Dave M, Elmunzer BJ, Dwamena BA, et al. Primary sclerosing cholangitis: meta-analysis of diagnostic performance of MR cholangiopancreatography. Radiology. 2010 Aug;256(2):387-96.
http://www.ncbi.nlm.nih.gov/pubmed/20656832?tool=bestpractice.com
[36]de Rougemont O, Dutkowski P, Clavien PA. Clinical update on inflammatory disorders of the GI tract: liver transplantation. In: Mayerle J, Tilg H, eds. Clinical update on inflammatory disorders of the gastrointestinal tract. Frontiers of Gastrointestinal Research. Vol. 26. Basel: Karger; 2010:59-71. MRCP can also be used to investigate the features of autoimmune hepatitis in children with PSC and to look for PSC in children with autoimmune hepatitis.[2]European Association for the Study of the Liver. EASL clinical practice guidelines on sclerosing cholangitis. J Hepatol. 2022 Sep;77(3):761-806.
https://www.doi.org/10.1016/j.jhep.2022.05.011
http://www.ncbi.nlm.nih.gov/pubmed/35738507?tool=bestpractice.com
ERCP should be avoided for the diagnosis of PSC.[3]Bowlus CL, Arrivé L, Bergquist A, et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2023 Feb 1;77(2):659-702.
https://www.doi.org/10.1002/hep.32771
http://www.ncbi.nlm.nih.gov/pubmed/36083140?tool=bestpractice.com
A 3D magnetic resonance imaging (MRI) MRCP with T1- and T2-weighted axial images and contrast enhancement should be obtained.[3]Bowlus CL, Arrivé L, Bergquist A, et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2023 Feb 1;77(2):659-702.
https://www.doi.org/10.1002/hep.32771
http://www.ncbi.nlm.nih.gov/pubmed/36083140?tool=bestpractice.com
The characteristic cholangiographic features of PSC are minor ductal irregularities, decreased branching or narrowing of the intrahepatic biliary tree, and multifocal stricturing of the intra- and extrahepatic bile ducts with areas of both normal caliber and dilated bile ducts, resulting in a "beaded" appearance. However, decreased branching or "diminution" of the intrahepatic biliary tree may also be present in patients with cirrhosis of any cause.[37]Terada T, Nakanuma Y. Intrahepatic cholangiographic appearance simulating primary sclerosing cholangitis in several hepatobiliary diseases: a postmortem cholangiographic and histopathological study in 154 livers at autopsy. Hepatology. 1995 Jul;22(1):75-81.
http://www.ncbi.nlm.nih.gov/pubmed/7601436?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Typical endoscopic retrograde cholangiopancreatography findings in a patient with PSC: multifocal strictures of the intra- and extrahepatic bile ductsDr Kris Kowdley's collection [Citation ends].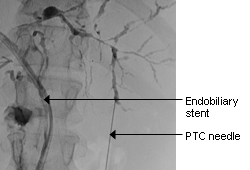 [Figure caption and citation for the preceding image starts]: Normal endoscopic retrograde cholangiopancreatographyDr Michael Saunders' collection [Citation ends].
[Figure caption and citation for the preceding image starts]: Normal endoscopic retrograde cholangiopancreatographyDr Michael Saunders' collection [Citation ends].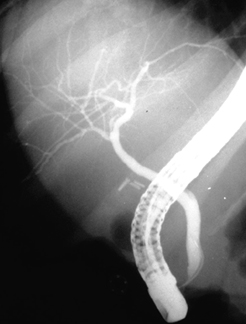 [Figure caption and citation for the preceding image starts]: Normal cholangiogram-pancreatogramDr Michael Saunders' collection [Citation ends].
[Figure caption and citation for the preceding image starts]: Normal cholangiogram-pancreatogramDr Michael Saunders' collection [Citation ends].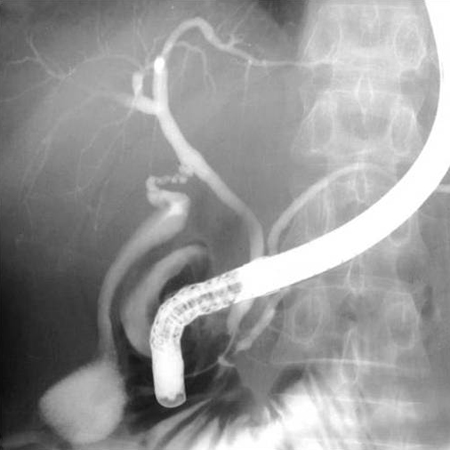
ERCP is indicated when MRCP is nondiagnostic or when therapeutic intervention is anticipated (e.g., brush cytology to evaluate for coexisting malignancy, extraction of bile duct stones, and dilation of prominent bile duct strictures).[2]European Association for the Study of the Liver. EASL clinical practice guidelines on sclerosing cholangitis. J Hepatol. 2022 Sep;77(3):761-806.
https://www.doi.org/10.1016/j.jhep.2022.05.011
http://www.ncbi.nlm.nih.gov/pubmed/35738507?tool=bestpractice.com
[3]Bowlus CL, Arrivé L, Bergquist A, et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2023 Feb 1;77(2):659-702.
https://www.doi.org/10.1002/hep.32771
http://www.ncbi.nlm.nih.gov/pubmed/36083140?tool=bestpractice.com
[30]Chapman R, Fevery J, Kalloo A, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010 Feb;51(2):660-78.
https://aasldpubs.onlinelibrary.wiley.com/doi/full/10.1002/hep.23294
http://www.ncbi.nlm.nih.gov/pubmed/20101749?tool=bestpractice.com
[35]Dave M, Elmunzer BJ, Dwamena BA, et al. Primary sclerosing cholangitis: meta-analysis of diagnostic performance of MR cholangiopancreatography. Radiology. 2010 Aug;256(2):387-96.
http://www.ncbi.nlm.nih.gov/pubmed/20656832?tool=bestpractice.com
[36]de Rougemont O, Dutkowski P, Clavien PA. Clinical update on inflammatory disorders of the GI tract: liver transplantation. In: Mayerle J, Tilg H, eds. Clinical update on inflammatory disorders of the gastrointestinal tract. Frontiers of Gastrointestinal Research. Vol. 26. Basel: Karger; 2010:59-71. Therapeutic ERCP should also be performed in patients with PSC if they have bacterial cholangitis that does not respond adequately to antibiotics.[2]European Association for the Study of the Liver. EASL clinical practice guidelines on sclerosing cholangitis. J Hepatol. 2022 Sep;77(3):761-806.
https://www.doi.org/10.1016/j.jhep.2022.05.011
http://www.ncbi.nlm.nih.gov/pubmed/35738507?tool=bestpractice.com
[3]Bowlus CL, Arrivé L, Bergquist A, et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2023 Feb 1;77(2):659-702.
https://www.doi.org/10.1002/hep.32771
http://www.ncbi.nlm.nih.gov/pubmed/36083140?tool=bestpractice.com
By definition, "dominant stricture" is a stenosis with a diameter of ≤1.5 mm in the common bile duct or with a diameter of ≤1 mm in the hepatic duct by ERCP. The term "dominant stricture" is used only with ERCP and not with MRI/MRCP, as MRI/MRCP has suboptimal spatial resolution. Common bile duct and hepatic duct strictures observed on MRI are termed "high-grade" or "severe" strictures.[2]European Association for the Study of the Liver. EASL clinical practice guidelines on sclerosing cholangitis. J Hepatol. 2022 Sep;77(3):761-806.
https://www.doi.org/10.1016/j.jhep.2022.05.011
http://www.ncbi.nlm.nih.gov/pubmed/35738507?tool=bestpractice.com
[3]Bowlus CL, Arrivé L, Bergquist A, et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2023 Feb 1;77(2):659-702.
https://www.doi.org/10.1002/hep.32771
http://www.ncbi.nlm.nih.gov/pubmed/36083140?tool=bestpractice.com
The term "relevant stricture" is used to describe strictures of clinical relevance not meeting the "dominant" and "high-grade" criteria. The European Association for the Study of the Liver (EASL) defines "relevant stricture" as a high-grade biliary stricture on imaging in the common bile duct or hepatic ducts with signs or symptoms of obstructive cholestasis and/or bacterial cholangitis (where a high-grade stricture is a biliary stricture on MRI/MRCP with >75% reduction of duct diameter in the common bile duct or hepatic ducts).[2]European Association for the Study of the Liver. EASL clinical practice guidelines on sclerosing cholangitis. J Hepatol. 2022 Sep;77(3):761-806.
https://www.doi.org/10.1016/j.jhep.2022.05.011
http://www.ncbi.nlm.nih.gov/pubmed/35738507?tool=bestpractice.com
The American Association for the Study of Liver Diseases (AASLD) defines "relevant stricture" as any biliary stricture of the common bile duct or hepatic duct associated with signs or symptoms of obstructive cholestasis and/or bacterial cholangitis.[3]Bowlus CL, Arrivé L, Bergquist A, et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2023 Feb 1;77(2):659-702.
https://www.doi.org/10.1002/hep.32771
http://www.ncbi.nlm.nih.gov/pubmed/36083140?tool=bestpractice.com
Bone mineral density scan
Bone mineral density scanning using dual energy x-ray absorptiometry (DEXA) is suggested in all patients at diagnosis and at 2- to 3- or 4-year intervals (based on risk factors) to detect osteoporosis.[2]European Association for the Study of the Liver. EASL clinical practice guidelines on sclerosing cholangitis. J Hepatol. 2022 Sep;77(3):761-806.
https://www.doi.org/10.1016/j.jhep.2022.05.011
http://www.ncbi.nlm.nih.gov/pubmed/35738507?tool=bestpractice.com
[3]Bowlus CL, Arrivé L, Bergquist A, et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2023 Feb 1;77(2):659-702.
https://www.doi.org/10.1002/hep.32771
http://www.ncbi.nlm.nih.gov/pubmed/36083140?tool=bestpractice.com
[30]Chapman R, Fevery J, Kalloo A, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010 Feb;51(2):660-78.
https://aasldpubs.onlinelibrary.wiley.com/doi/full/10.1002/hep.23294
http://www.ncbi.nlm.nih.gov/pubmed/20101749?tool=bestpractice.com
[34]Lindor KD, Kowdley KV, Harrison ME; American College of Gastroenterology. ACG clinical guideline: primary sclerosing cholangitis. Am J Gastroenterol. 2015 May;110(5):646-59.
https://gi.org/guideline/primary-sclerosing-cholangitis
http://www.ncbi.nlm.nih.gov/pubmed/25869391?tool=bestpractice.com
[38]Zein CO, Jorgensen RA, Clarke B, et al. Alendronate improves bone mineral density in primary biliary cirrhosis: a randomized placebo-controlled trial. Hepatology. 2005 Oct;42(4):762-71.
https://aasldpubs.onlinelibrary.wiley.com/doi/full/10.1002/hep.20866
http://www.ncbi.nlm.nih.gov/pubmed/16175618?tool=bestpractice.com
[39]Collier J. Bone disorders in chronic liver disease. Hepatology. 2007 Oct;46(4):1271-8.
https://aasldpubs.onlinelibrary.wiley.com/doi/full/10.1002/hep.21852
http://www.ncbi.nlm.nih.gov/pubmed/17886334?tool=bestpractice.com
Colonoscopy
Patients newly diagnosed with PSC should undergo ileocolonoscopy with biopsies regardless of whether or not they already have a diagnosis of inflammatory bowel disease.
Ileocolonoscopy should be repeated (US guidance) or can be considered (European guidelines) every 5 years if inflammatory bowel disease is not present or whenever the patient has symptoms suggestive of inflammatory bowel disease.[2]European Association for the Study of the Liver. EASL clinical practice guidelines on sclerosing cholangitis. J Hepatol. 2022 Sep;77(3):761-806.
https://www.doi.org/10.1016/j.jhep.2022.05.011
http://www.ncbi.nlm.nih.gov/pubmed/35738507?tool=bestpractice.com
[3]Bowlus CL, Arrivé L, Bergquist A, et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2023 Feb 1;77(2):659-702.
https://www.doi.org/10.1002/hep.32771
http://www.ncbi.nlm.nih.gov/pubmed/36083140?tool=bestpractice.com
From the age of 15 years, all patients with coexisting inflammatory bowel disease should undergo high-definition surveillance colonoscopy regularly due to a high risk of colorectal cancer. US guidance suggests 1- to 2-year surveillance intervals, whereas European guidelines recommend surveillance annually, with 1- to 2-year intervals if there is no inflammatory activity.[2]European Association for the Study of the Liver. EASL clinical practice guidelines on sclerosing cholangitis. J Hepatol. 2022 Sep;77(3):761-806.
https://www.doi.org/10.1016/j.jhep.2022.05.011
http://www.ncbi.nlm.nih.gov/pubmed/35738507?tool=bestpractice.com
[3]Bowlus CL, Arrivé L, Bergquist A, et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2023 Feb 1;77(2):659-702.
https://www.doi.org/10.1002/hep.32771
http://www.ncbi.nlm.nih.gov/pubmed/36083140?tool=bestpractice.com
[30]Chapman R, Fevery J, Kalloo A, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010 Feb;51(2):660-78.
https://aasldpubs.onlinelibrary.wiley.com/doi/full/10.1002/hep.23294
http://www.ncbi.nlm.nih.gov/pubmed/20101749?tool=bestpractice.com
[34]Lindor KD, Kowdley KV, Harrison ME; American College of Gastroenterology. ACG clinical guideline: primary sclerosing cholangitis. Am J Gastroenterol. 2015 May;110(5):646-59.
https://gi.org/guideline/primary-sclerosing-cholangitis
http://www.ncbi.nlm.nih.gov/pubmed/25869391?tool=bestpractice.com
[40]Vleggaar FP, Lutgens MW, Claessen MM, et al. Review article: the relevance of surveillance endoscopy in long-lasting inflammatory bowel disease. Aliment Pharmacol Ther. 2007 Dec;26 Suppl 2:47-52.
http://www.ncbi.nlm.nih.gov/pubmed/18081648?tool=bestpractice.com
Staging of fibrosis
The AASLD suggests using imaging-based noninvasive testing to detect advanced fibrosis and cirrhosis in adults with PSC.[41]Sterling RK, Duarte-Rojo A, Patel K, et al. AASLD practice guideline on imaging-based noninvasive liver disease assessment of hepatic fibrosis and steatosis. Hepatology. 2025 Feb 1;81(2):672-724.
https://journals.lww.com/hep/fulltext/2025/02000/aasld_practice_guideline_on_imaging_based.30.aspx
Either transient elastography or magnetic resonance elastography is recommended to stage fibrosis.[3]Bowlus CL, Arrivé L, Bergquist A, et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2023 Feb 1;77(2):659-702.
https://www.doi.org/10.1002/hep.32771
http://www.ncbi.nlm.nih.gov/pubmed/36083140?tool=bestpractice.com
[41]Sterling RK, Duarte-Rojo A, Patel K, et al. AASLD practice guideline on imaging-based noninvasive liver disease assessment of hepatic fibrosis and steatosis. Hepatology. 2025 Feb 1;81(2):672-724.
https://journals.lww.com/hep/fulltext/2025/02000/aasld_practice_guideline_on_imaging_based.30.aspx
The AASLD advises against using imaging-based tests as standalone tests to assess regression or progression of liver fibrosis.[41]Sterling RK, Duarte-Rojo A, Patel K, et al. AASLD practice guideline on imaging-based noninvasive liver disease assessment of hepatic fibrosis and steatosis. Hepatology. 2025 Feb 1;81(2):672-724.
https://journals.lww.com/hep/fulltext/2025/02000/aasld_practice_guideline_on_imaging_based.30.aspx
Histology
Liver biopsy may support the diagnosis of PSC but is rarely diagnostic and is not necessary if clinical, laboratory, and radiographic findings already suggest PSC. There is evidence to suggest that liver biopsy in patients with cholangiographically diagnosed PSC adds new information or affects management in only 1.3% of patients.[42]Burak KW, Angulo P, Lindor KD. Is there a role for liver biopsy in primary sclerosing cholangitis? Am J Gastroenterol. 2003 May;98(5):1155-8.
http://www.ncbi.nlm.nih.gov/pubmed/12809842?tool=bestpractice.com
However, liver biopsy should be considered (US guidance) and is recommended (European guidelines) to diagnose small-duct PSC (which should be suspected with, and is characterized by, laboratory and histologic findings typical for PSC but with normal cholangiogram, especially in the presence of inflammatory bowel disease) or to evaluate for alternative or overlapping disorders, such as PSC-autoimmune hepatitis.[2]European Association for the Study of the Liver. EASL clinical practice guidelines on sclerosing cholangitis. J Hepatol. 2022 Sep;77(3):761-806.
https://www.doi.org/10.1016/j.jhep.2022.05.011
http://www.ncbi.nlm.nih.gov/pubmed/35738507?tool=bestpractice.com
[3]Bowlus CL, Arrivé L, Bergquist A, et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2023 Feb 1;77(2):659-702.
https://www.doi.org/10.1002/hep.32771
http://www.ncbi.nlm.nih.gov/pubmed/36083140?tool=bestpractice.com
[34]Lindor KD, Kowdley KV, Harrison ME; American College of Gastroenterology. ACG clinical guideline: primary sclerosing cholangitis. Am J Gastroenterol. 2015 May;110(5):646-59.
https://gi.org/guideline/primary-sclerosing-cholangitis
http://www.ncbi.nlm.nih.gov/pubmed/25869391?tool=bestpractice.com
Liver biopsy is also useful for staging disease (primarily for detecting cirrhosis, if not clinically apparent).[30]Chapman R, Fevery J, Kalloo A, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010 Feb;51(2):660-78.
https://aasldpubs.onlinelibrary.wiley.com/doi/full/10.1002/hep.23294
http://www.ncbi.nlm.nih.gov/pubmed/20101749?tool=bestpractice.com
The pathognomonic histologic feature of PSC is fibro-obliterative cholangitis, which is found in <12% of patients with PSC who undergo liver biopsy.[43]Wiesner RH, LaRusso NF, Ludwig J, et al. Comparison of the clinicopathologic features of primary sclerosing cholangitis and primary biliary cirrhosis. Gastroenterology. 1985 Jan;88(1 Pt 1):108-14.
http://www.ncbi.nlm.nih.gov/pubmed/3880553?tool=bestpractice.com
Other histologic findings are nonspecific and include: periductal fibrosis and inflammation, absence of bile ducts in some portal tracts (referred to as focal ductopenia), and ductal proliferation in other portal tracts.[44]Ludwig J, Barham SS, LaRusso NF, et al. Morphologic features of chronic hepatitis associated with primary sclerosing cholangitis and chronic ulcerative colitis. Hepatology. 1981 Nov-Dec;1(6):632-40.
http://www.ncbi.nlm.nih.gov/pubmed/7308996?tool=bestpractice.com
During interventional procedures accessing the biliary tract, brush cytology should be performed to rule out cholangiocarcinoma, although yields are notably as low as 18%.[45]Baron TH, Harewood GC, Rumalla A, et al. A prospective comparison of digital image analysis and routine cytology for the identification of malignancy in biliary tract strictures. Clin Gastroenterol Hepatol. 2004 Mar;2(3):214-9.
http://www.ncbi.nlm.nih.gov/pubmed/15017605?tool=bestpractice.com
Diagnosis of cholangiocarcinoma remains difficult, and workup should include tumor markers such as CA 19-9, imaging, and fluorescence in situ hybridization (FISH) analysis of intraductal tissue samples obtained during ERCP.[2]European Association for the Study of the Liver. EASL clinical practice guidelines on sclerosing cholangitis. J Hepatol. 2022 Sep;77(3):761-806.
https://www.doi.org/10.1016/j.jhep.2022.05.011
http://www.ncbi.nlm.nih.gov/pubmed/35738507?tool=bestpractice.com
[3]Bowlus CL, Arrivé L, Bergquist A, et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2023 Feb 1;77(2):659-702.
https://www.doi.org/10.1002/hep.32771
http://www.ncbi.nlm.nih.gov/pubmed/36083140?tool=bestpractice.com
[30]Chapman R, Fevery J, Kalloo A, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010 Feb;51(2):660-78.
https://aasldpubs.onlinelibrary.wiley.com/doi/full/10.1002/hep.23294
http://www.ncbi.nlm.nih.gov/pubmed/20101749?tool=bestpractice.com
[46]Liew ZH, Loh TJ, Lim TK, et al. Role of fluorescence in situ hybridization in diagnosing cholangiocarcinoma in indeterminate biliary strictures. J Gastroenterol Hepatol. 2018 Jan;33(1):315-9.
http://www.ncbi.nlm.nih.gov/pubmed/28543841?tool=bestpractice.com