Approach
An accurate diagnosis requires a detailed description of the clinical episodes and careful analysis of the electroencephalogram (EEG).
History
As is true for all epilepsies, a detailed eye witness description is imperative. Home video recordings can be invaluable, and direct observation of seizures in the clinical setting may be possible as clusters tend to occur frequently throughout the day.
The spasms are usually symmetric, bilateral, brief contractions of the axial muscle groups, and may involve the flexors, extensors, or both. The flexor spasm is most common; however, presentation may be subtle, merely with head nodding or eyelid fluttering. Early in the disease course, spasms may be isolated events with the tendency to cluster developing later. Clusters of up to 50-100 serial spasms may recur frequently over 24 hours. Spasms may be precipitated by sleep onset/offset or feeding, frequently leading to misdiagnosis as gastroesophageal reflux or infantile colic.
Typical age of onset is between 1 month and 1 year of age, with a peak age of onset between 3 and 5 months. Some may present as late as 3 years. There may be a preceding history of developmental delay, which may be consistent with the underlying etiology (e.g., Down syndrome). However, onset of spasms is often associated with plateauing or regression of development, even in an infant previously considered to be developing normally. At presentation, careful attention should be given to the child’s past and present developmental status.
A detailed prenatal and birth history may suggest significant perinatal insults to the developing brain, such as hypoxia, prenatal or neonatal stroke, or sinovenous thrombosis. Hiccups, excessive startle, feeding difficulties, and faltering growth may indicate a potential metabolic etiology. Careful attention should be given to family history that may suggest an underlying etiology (e.g., tuberous sclerosis). There is often a family history of other epilepsy syndromes.
Exam
A careful general exam should identify dysmorphology and neurocutaneous stigmata associated with common genetic syndromes such as Down syndrome and tuberous sclerosis. A Wood lamp exam is imperative, as ash-leaf macules typical of tuberous sclerosis may otherwise be unrecognized. The lesions fluoresce under ultraviolet light. [Figure caption and citation for the preceding image starts]: Ash-leaf macules in a child with tuberous sclerosisFrom the collection of Dr Teesta Soman and Dr Shelly Weiss [Citation ends].
Growth parameters, including head circumference, should be plotted accurately on appropriate charts. Microcephaly at presentation may suggest structural brain malformations, perinatal injuries, vascular events, inherited metabolic disorders, or intrauterine infections.
Examination of tone and movement patterns may reveal truncal hypotonia or peripheral spasticity. Asymmetry or focal deficits on exam may be suggestive of cortical malformations, space-occupying lesions, infection, or preceding injury to the developing brain. However, at presentation, the baseline neurologic exam may be normal and uninformative.
Cardiovascular exam may identify comorbidities of tuberous sclerosis or genetic syndromes such as Down syndrome. Ophthalmologic exam may suggest an underlying syndrome or perinatal infection (e.g., cytomegalovirus or toxoplasmosis).
It is important to record the premorbid developmental status at presentation because the infant may be developmentally age-appropriate or have features of developmental delay. Delay may occur in all developmental areas, including fine motor, gross motor, speech and language, and vision.
EEG
An EEG should be obtained in all patients as soon as the diagnosis is suspected. EEG typically, although not universally, reveals hypsarrhythmia.[26]
[Figure caption and citation for the preceding image starts]: EEG demonstrating hypsarrhythmiaFrom the collection of Dr Teesta Soman and Dr Shelly Weiss [Citation ends].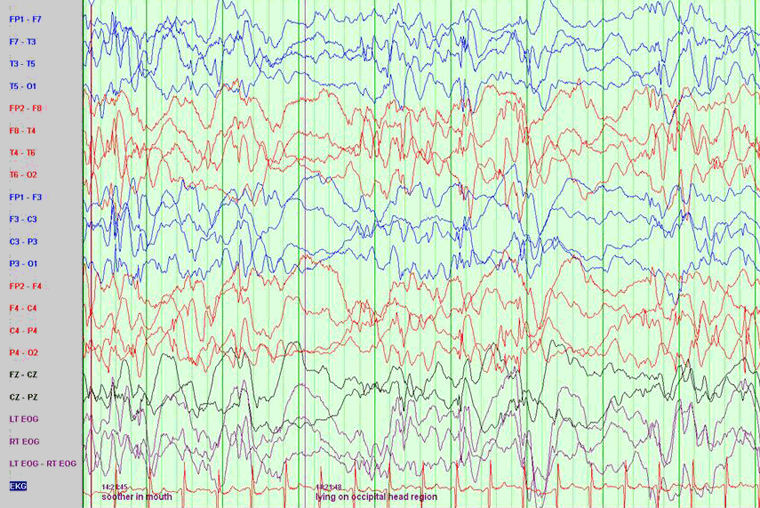
Hypsarrhythmia is characterized by random high-voltage spike and slow waves of varying amplitude, arising from multiple foci that vary over time. The background is asynchronous and generally chaotic.[27] Typical hypsarrhythmia is more likely in the early phase of infantile spasms and in younger patients.[26] It may evolve over time into multifocal and interictal epileptic discharges. The interictal EEG is very abnormal in this syndrome but occasionally is only abnormal during sleep. Hypsarrhythmia may disappear during REM sleep and is found with greater sensitivity during other sleep stages.
If there is a strong clinical suspicion of infantile spasms, a prolonged sleep recording is required as the typical hypsarrhythmic pattern may be missed.[26]
Hypsarrhythmia may be asymmetric, suggesting a lesional epilepsy, cortical malformation, or disorder of neuronal migration. Modified hypsarrhythmia may be more synchronous, less symmetric, and show more focal changes or areas of attenuation. Interictal (multi) focal discharges can also be seen on EEG.
The ictal EEG is variable but may show suppression, fast activity, or attenuation of the background activity (an electrodecremental response), which may be preceded by high-amplitude vertex slow waves or spindle-like activity.
Imaging
Magnetic resonance imaging (MRI) of the brain may identify underlying structural and migrational abnormalities. The scan may show areas of malformation or lesions secondary to hemorrhage, infection, infarction, or tumor (e.g., calcification, porencephaly [cyst or cavity in cerebral hemisphere], and encephalomalacia [softening of the brain tissue]). Stigmata of tuberous sclerosis may be observed, including cortical hamartomas, subependymal nodules, and subependymal giant cell astrocytomas. Characteristic abnormal patterns may suggest neurometabolic or neurocutaneous syndromes. However, neuroimaging may be entirely normal.
A computed tomography brain scan is less sensitive and should be considered only if an MRI scan is not initially easily obtained.[Figure caption and citation for the preceding image starts]: Cortical tubers on MRI in tuberous sclerosisFrom the collection of Dr Teesta Soman and Dr Shelly Weiss [Citation ends].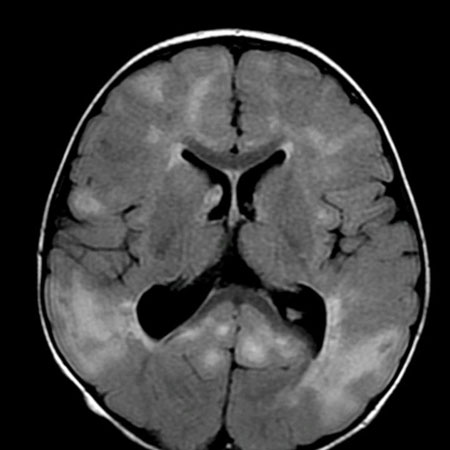
Genetic testing
If available, and if no clear underlying etiology is found after clinical exam and MRI, next-generation sequencing genetic tests (gene panel/whole-exome sequencing/whole-genome sequencing) should be considered early.[28][29][30] Next-generation sequencing should also be considered for patients with structural brain disorders known to be associated with genetic mutations such as tuberous sclerosis. Microarray comparative genome hybridization (CGH) analysis is also recommended in most patients, as chromosomal abnormalities and copy number variants have been associated with infantile spasms. If microarray CGH is not available, chromosomal analysis should be performed.
A genetic etiology can be defined in up to 41% of cases of infantile spasms, including trisomy 21, CDKL5, STXBP1, ARX, IQSEC2, TSC1, TSC2, and more.[30][31][32] A genetic mutation can be de novo or inherited from a parent with mild symptoms or an unaffected parent. Next-generation sequencing genetic tests are preferably performed as a trio analysis, meaning both biological parents’ and childs’ samples are taken and analyzed simultaneously.
If there are existing genetic test results for a patient, do not perform repeat testing unless there is uncertainty about the existing result, for example, the result is inconsistent with the patient’s clinical presentation or the test methodology has changed.[33]
Metabolic and other laboratory investigations
Besides genetic testing, initial tests at presentation should include:
Complete blood count
Blood chemistries
Glucose
Serum calcium and magnesium
Liver function tests
Serum ammonia
Blood gas/bicarbonate
Serum lactate/pyruvate
Plasma amino acids
Biotinidase
Urine organic acids
Acylcarnitines
Urine and plasma creatine and guanidinoacetate
Urine alpha-amino adipic semialdehyde dehydrogenase (AASA)
Cerebrospinal fluid (CSF) exam: this should include CSF glucose and lactate paired with plasma, amino acids, pyridoxal-5-phosphate, and methyltetrahydrofolate.
Subsequently, the following tests can be considered:
Thyroid function tests (free thyroxine [FT4], thyroid-stimulating hormone [TSH])
Plasma transferrin glycoforms
Very long-chain fatty acids
Urine sulfocysteine.
Other tests to consider if indicated:
A screen for intrauterine infections including cytomegalovirus (CMV) and toxoplasmosis (toxoplasma and CMV antibodies, and CMV polymerase chain reaction [PCR] in urine)
Echocardiogram and renal ultrasound scan if there is clinical or radiologic evidence of tuberous sclerosis
Ophthalmic review for evidence of chorioretinitis in intrauterine infections.
Use of this content is subject to our disclaimer