Etiology
Physiologic jaundice can be a result of:
Increased bilirubin load secondary to increased red blood cell (RBC) volume, decreased RBC life span, or increased enterohepatic circulation
Decreased uptake by the liver because of decreased ligandins or binding of ligandins to other anions
Decreased conjugation in the liver because of decreased uridine diphosphoglucuronyl transferase (UDPGT) activity. UGT1A1 gene polymorphisms of Gly71Arg and TATA promoter, which decrease UDPGT enzymatic activity, have been noted to be significant risk factors associated with neonatal hyperbilirubinemia.[9]
Decreased excretion into bile.
Pathologic jaundice with unconjugated hyperbilirubinemia can be a result of:
Hemolytic anemias: these result in increased destruction of RBCs, with resultant increased heme, which is converted to excess unconjugated bilirubin; the immature liver is unable to handle the excess load. They can be the result of blood group incompatibility (rhesus, ABO), RBC enzyme defects (glucose-6-phosphate dehydrogenase deficiency; pyruvate kinase deficiency), RBC membrane defects (e.g., hereditary spherocytosis, infantile pyknocytosis), thalassemia, drug-induced (by vitamin K, sulfonamides, nitrofurantoin, anti-malarials, penicillin), or sepsis
Extravasation of blood: sequestration of blood in cavities results in increased bilirubin load. Examples include cephalhematoma; intracranial, pulmonary, or gastrointestinal hemorrhage; large hemangiomas; excessive ecchymoses; or petechiae
Polycythemia: increased number of RBCs leads to increased production of bilirubin
Increased enterohepatic circulation: delayed gastrointestinal transit increases bilirubin levels. Examples include intestinal atresia/stenosis, pyloric stenosis, Hirschsprung disease, meconium ileus/plug syndrome
Defective conjugation: congenital deficiencies of UDPGT enzyme include Crigler-Najjar syndrome; UDPGT enzyme inhibition can be the result of drugs (e.g., novobiocin) or Lucey-Driscoll syndrome
Metabolic conditions (galactosemia, hypothyroidism, tyrosinosis, hypermethioninemia, maternal diabetes)
Breast-feeding: “suboptimal intake hyperbilirubinemia” associated with inadequate breast milk intake typically peaks on days 3 to 5 after birth and is frequently associated with excess weight loss. Decreased stool frequency leads to an increased enterohepatic circulation of bilirubin. “Breast milk jaundice” or the “breast milk jaundice syndrome,” in contrast, persists up to 3 months despite adequate human milk intake and optimal weight gain.[7]
Decreased binding of bilirubin to albumin: increased availability of the free (unconjugated) bilirubin to cross the blood-brain barrier. This can be caused by drugs (sulfonamides, penicillin, gentamicin), acidosis, asphyxia, hypothermia, increased osmolality, or hypoglycemia.
Pathologic jaundice with conjugated hyperbilirubinemia (direct bilirubin is >2.0 mg/dL) can be a result of:
Hepatocellular disease:
Metabolic or genetic defects. Examples include alpha1-antitrypsin deficiency, cystic fibrosis, Zellweger syndrome, Dubin-Johnson syndrome (absence of multidrug resistance-associated protein 2 [MRP2] from the canalicular membrane of hepatocytes), Rotor syndrome (organic-anion-transporting polypeptide [OATP]1B1 and OATP1B3 are absent at the sinusoidal membrane of hepatocytes), and galactosemia
Infections. Examples include rubella, cytomegalovirus, herpes, syphilis, hepatitis A and B, toxoplasmosis, and urinary tract infection with Escherichia coli
Total parenteral nutrition[10]
Neonatal hemochromatosis
Idiopathic neonatal hepatitis
Shock.
Intrahepatic biliary disease because of Alagille syndrome (arteriohepatic dysplasia), or inspissated bile syndrome
Extrahepatic biliary disease because of biliary atresia, choledochal cyst, bile duct stenosis, cholelithiasis.
Pathophysiology
Bilirubin is the final product of heme catabolism, which is mostly derived from hemoglobin. Breakdown of red blood cell hemoglobin results in heme production (75% of source of bilirubin). Heme can also be derived (25% of source of bilirubin) from breakdown of other proteins such as myoglobin, cytochromes, and nitric oxide synthases. In the reticulo-endothelial system, heme is further catabolized by heme oxygenase (rate-limiting step for bilirubin production) to biliverdin. This is acted upon by biliverdin reductase to form bilirubin. Bilirubin is then bound to serum albumin in the plasma and is transported to the liver. Bilirubin dissociates from albumin, and with the help of carrier proteins such as ligandins, is taken up into the hepatocytes. Bilirubin is then conjugated by the uridine diphosphoglucuronyl transferase enzyme in the hepatocytes. Bilirubin glucuronide reaches the intestines via the gallbladder and common bile duct. In the intestines of the newborn, most of the bilirubin glucuronide is unconjugated by beta glucuronidase. Some of this unconjugated bilirubin is reabsorbed and gets into the enterohepatic circulation. The rest of the conjugated bilirubin reaches the colon, where the bacteria break it down to urobilinogen, which is then excreted. When this normal process of bilirubin formation and excretion is disrupted, hyperbilirubinemia results.[11]
[Figure caption and citation for the preceding image starts]: Metabolic pathway of bilirubin with pathologies relating to unconjugated hyperbilirubinemia. 1. ABO incompatibility, Rhesus incompatibility, shorter RBC lifespan in neonates, bruising during delivery; 2. induced by inflammatory mediators associated with comorbidities of prematurity (e.g., respiratory distress syndrome, infection); 3. dissociation increased by acidosis, ketosis, renal failure; 4. permeable blood-brain barrier in term neonates and premature babies; 5. mutation in UGT1A1 gene results in Gilbert syndrome or Crigler-Najjar syndrome I and II; 6. unconjugated bilirubin load increased by decreased gut motilityCreated by BMJ Knowledge Centre [Citation ends].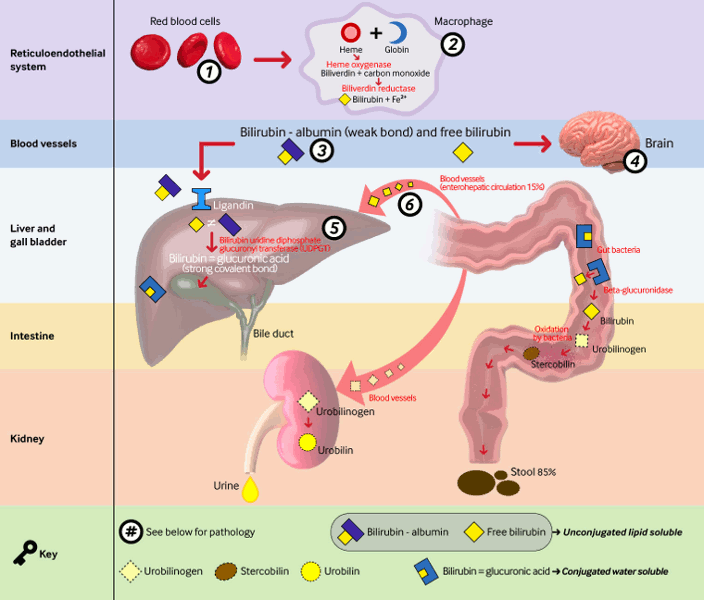 [Figure caption and citation for the preceding image starts]: Detail of liver lobule and its functions, highlighting pathologies that cause conjugated hyperbilirubinemia. Pathologies include: 1. portal vein thrombosis; 2. choledochal cyst; 3. infection (sepsis, E coli urinary tract infection, hepatitis A or B, toxoplasmosis, cytomegalovirus, syphilis, herpes), metabolic (Rotor syndrome, galactosemia, tyrosinemia, alpha-1 antitrypsin deficiency, hypothyroidism, cystic fibrosis, Zellweger syndrome), drugs, idiopathic neonatal hepatitis, total parenteral nutrition, neonatal hemochromatosis, shock/hypoxia/ischemia; 4. bile duct paucity, biliary atresia, Alagille syndrome, idiopathic neonatal cholestasis, progressive familial intrahepatic cholestasis, inspissated bile syndrome; 5. MRP2 (also known as ABCC2) gene mutations on the canalicular membrane of hepatocytes result in Dubin-Johnson syndrome, absence of OATP1B1 and OATP1B3 at the sinusoidal membrane of hepatocytes result in Rotor syndromeCreated by BMJ Knowledge Centre [Citation ends].
[Figure caption and citation for the preceding image starts]: Detail of liver lobule and its functions, highlighting pathologies that cause conjugated hyperbilirubinemia. Pathologies include: 1. portal vein thrombosis; 2. choledochal cyst; 3. infection (sepsis, E coli urinary tract infection, hepatitis A or B, toxoplasmosis, cytomegalovirus, syphilis, herpes), metabolic (Rotor syndrome, galactosemia, tyrosinemia, alpha-1 antitrypsin deficiency, hypothyroidism, cystic fibrosis, Zellweger syndrome), drugs, idiopathic neonatal hepatitis, total parenteral nutrition, neonatal hemochromatosis, shock/hypoxia/ischemia; 4. bile duct paucity, biliary atresia, Alagille syndrome, idiopathic neonatal cholestasis, progressive familial intrahepatic cholestasis, inspissated bile syndrome; 5. MRP2 (also known as ABCC2) gene mutations on the canalicular membrane of hepatocytes result in Dubin-Johnson syndrome, absence of OATP1B1 and OATP1B3 at the sinusoidal membrane of hepatocytes result in Rotor syndromeCreated by BMJ Knowledge Centre [Citation ends].
Classification
Physiologic jaundice
Physiologic jaundice is usually noted at postpartum day 2, peaks on days 3 through 5, and then decreases. Serum bilirubin levels up to 12 mg/dL are considered physiologic in term neonates.
Pathologic jaundice
Any jaundice in the first 24 hours of life is considered pathologic. Bilirubin levels exceeding 95th percentile, as defined by a nomogram, are pathologic.[1]
Use of this content is subject to our disclaimer