Etiology
Autoimmune subepidermal blistering skin diseases include the bullous pemphigoid group of diseases (bullous pemphigoid, pemphigoid gestationis, lichen planus pemphigoides, linear IgA disease, cicatricial pemphigoid, and anti-p200, anti-p105, and anti-p450 pemphigoid), epidermolysis bullosa acquisita, and dermatitis herpetiformis. With the exception of dermatitis herpetiformis, all these disorders are characterized by circulating and tissue-bound autoantibodies against various components of the dermal-epidermal anchoring complex,[15] which is a supramolecular structure that mediates the adhesion of basal keratinocytes to the underlying dermis. Antibody binding to various proteins within this complex results in dermal-epidermal separation and tense blister formation.
The dermal-epidermal anchoring complex consists of hemidesmosomes of the basal keratinocytes, anchoring filaments of the basement membrane, and anchoring fibrils of the papillary dermis. Structural proteins within this complex, described as autoantigens in various autoimmune bullous dermatoses, include bullous pemphigoid antigen 180 (bullous pemphigoid, pemphigoid gestationis, mucous membrane pemphigoid, linear IgA bullous dermatosis), BP230 (bullous pemphigoid), alpha 6 beta 4 integrin (mucous membrane pemphigoid), laminin 5 and 6 (mucous membrane pemphigoid), and type VII collagen (epidermolysis bullosa acquisita).[16] More than 300 distinct mutations in 10 different genes corresponding to structural components of the basement membrane have been described that result in skin fragility and dermal-epidermal separation associated with characteristic extracutaneous manifestations.[15]
Pathophysiology
Autoantibodies are directed against two hemidesmosomal proteins, designated BP180 and BP230. While BP230 localizes intracellularly and associates with the hemidesmosomal plaque, BP180 is a transmembrane glycoprotein with an extracellular domain consisting of approximately 1000 amino acids. The noncollagenous 16A domain that encompasses 76 amino acids and localizes directly adjacent to the transmembrane region has been identified as an immunodominant region of the BP180 ectodomain.[17]
In most bullous pemphigoid sera, circulating antibodies to BP180NC16A are detected with serum levels correlating with disease activity.[18] Bullous pemphigoid autoantibodies (with the help of complement activation), the infiltration of inflammatory cells, and the release of proteases and various inflammatory mediators, including cytokines, are essential for lesion formation.[19][20] Further studies using a similar approach revealed that antihuman BP180NC16A antibodies and neutrophils are responsible for this tissue injury.[21] Using an IgG passive transfer approach, there exists in vivo evidence that rabbit antibodies directed against the murine BP180NC16A homolog are pathogenic in mice.[22] These in vitro and in vivo data demonstrate that antibodies specific for the BP180NC16A domain are pathogenic. It is clear that the binding of anti-BP180 antibody to its target is the critical first step in subepidermal blister formation in bullous pemphigoid. It is unclear as to the subclass of IgG that is pathogenic and directly related to the severity of the disease. New findings identify anti-BP180-N IgG1 autoantibodies as the predominant IgG subclass associated with acute onset of bullous pemphigoid. Because IgG1 antibodies are capable of fixing complement, the link between anti-BP180 autoantibodies and complement in their pathogenic role in bullous pemphigoid is revealed.[1][23]
Bullous pemphigoid often provokes blood and tissue eosinophilia, which suggests chemoattractants may modulate the eosinophil infiltration. Eotaxin and interleukin (IL)-5 are strongly associated with the tissue eosinophilia of bullous pemphigoid. These findings suggest that eotaxin and IL-5 may be important for eosinophil migration in bullous pemphigoid lesions and that therapies that aim to inhibit production of eotaxin and IL-5 may improve inflammation and blister formation.[24]
Classification
Clinical classification
Several bullous pemphigoid clinical variants have been described:[2][3]
Classic (bullous): most common, tense bullae arise anywhere, oral and ocular involvement rare, minor, heal without scarring or milia[Figure caption and citation for the preceding image starts]: Tense, fluid-filled blisters on normal and erythematous skinFrom the collection of Dr Vesna Petronic-Rosic [Citation ends].
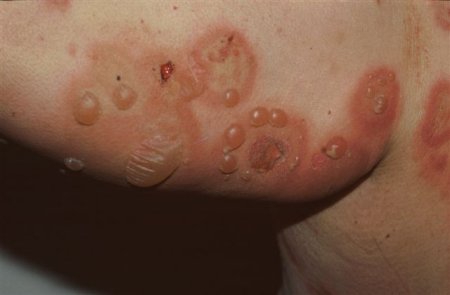 [Figure caption and citation for the preceding image starts]: Tense, fluid-filled blisters on normal and erythematous skin of the trunk and extremitiesFrom the collection of Dr Vesna Petronic-Rosic [Citation ends].
[Figure caption and citation for the preceding image starts]: Tense, fluid-filled blisters on normal and erythematous skin of the trunk and extremitiesFrom the collection of Dr Vesna Petronic-Rosic [Citation ends].
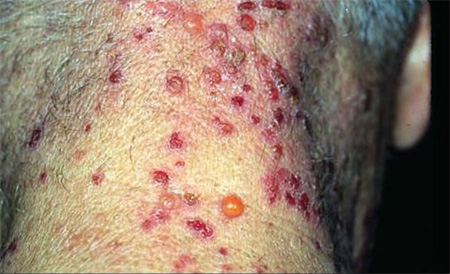
Vesicular: uncommon; groups of small, tense blisters on a red or itchy skin base[Figure caption and citation for the preceding image starts]: Vesicular variant: tense blistersFrom the collection of Dr Vesna Petronic-Rosic [Citation ends].
Nodular: rare, blisters on normal or nodular skin
Pretibial: bullae limited to one body area, such as the shins
Dyshidrosiform: blisters localized to palms and soles
Vegetative: very rare; vegetating plaques in the axilla, neck, and/or groin
Generalized: rare, may look like psoriasis or atopic dermatitis, may develop vesicles or bullae
Urticarial: initially presents with urticarial areas that become bullous lesions, some never develop bullae
Childhood: associated with vaccination; bullae on palms, soles, face
Erythrodermic: erythroderma seen first all over body, then eruption of isolated bullae.
Use of this content is subject to our disclaimer