The diagnostic workup of amyloidosis includes a detailed history and physical exam; laboratory and pathologic evaluation (including studies to confirm the presence and type of amyloid deposits in tissue); and imaging studies.
It is important to determine amyloidosis type when making a diagnosis, as this guides treatment.
History
A detailed history should be carried out to help to determine the potential cause and type of amyloidosis.
History may reveal a prior diagnosis of monoclonal gammopathy of undetermined significance (MGUS).[57]Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002 Feb 21;346(8):564-9.
https://www.doi.org/10.1056/NEJMoa01133202
http://www.ncbi.nlm.nih.gov/pubmed/11856795?tool=bestpractice.com
Patients with MGUS have a relative risk of progression to AL amyloidosis (the most common type of amyloidosis) of eight- to ninefold.[4]Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016 Jun 25;387(10038):2641-54.
http://www.ncbi.nlm.nih.gov/pubmed/26719234?tool=bestpractice.com
[57]Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002 Feb 21;346(8):564-9.
https://www.doi.org/10.1056/NEJMoa01133202
http://www.ncbi.nlm.nih.gov/pubmed/11856795?tool=bestpractice.com
[58]Kyle RA, Larson DR, Therneau TM, et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Engl J Med. 2018 Jan 18;378(3):241-9.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5852672
http://www.ncbi.nlm.nih.gov/pubmed/29342381?tool=bestpractice.com
It is important to note, however, that an incidental monoclonal gammopathy may be present in patients with other types of amyloidosis (e.g., hereditary amyloidosis, wild-type transthyretin [ATTRwt] amyloidosis), which can lead to a misdiagnosis of AL amyloidosis.[27]Comenzo RL, Zhou P, Fleisher M, et al. Seeking confidence in the diagnosis of systemic AL (Ig light-chain) amyloidosis: patients can have both monoclonal gammopathies and hereditary amyloid proteins. Blood. 2006 May 1;107(9):3489-91.
http://www.bloodjournal.org/content/107/9/3489.full
http://www.ncbi.nlm.nih.gov/pubmed/16439680?tool=bestpractice.com
[28]Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002 Jun 6;346(23):1786-91.
http://www.nejm.org/doi/full/10.1056/NEJMoa013354#t=article
http://www.ncbi.nlm.nih.gov/pubmed/12050338?tool=bestpractice.com
[60]Phull P, Sanchorawala V, Connors LH, et al. Monoclonal gammopathy of undetermined significance in systemic transthyretin amyloidosis (ATTR). Amyloid. 2018 Mar;25(1):62-7.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6157907
http://www.ncbi.nlm.nih.gov/pubmed/29424556?tool=bestpractice.com
Clinical awareness together with a careful family history and laboratory/pathologic evaluation (including genetic testing) are essential to avoid a misdiagnosis.[29]Gertz M, Adams D, Ando Y, et al. Avoiding misdiagnosis: expert consensus recommendations for the suspicion and diagnosis of transthyretin amyloidosis for the general practitioner. BMC Fam Pract. 2020 Sep 23;21(1):198.
https://www.doi.org/10.1186/s12875-020-01252-4
http://www.ncbi.nlm.nih.gov/pubmed/32967612?tool=bestpractice.com
[61]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
Secondary (AA) amyloidosis is associated with:[16]Lachmann HJ, Goodman HJB, Gilbertson JA, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007 Jun 7;356(23):2361-71.
https://www.nejm.org/doi/10.1056/NEJMoa070265
http://www.ncbi.nlm.nih.gov/pubmed/17554117?tool=bestpractice.com
[21]Fayand A, Boutboul D, Galicier L, et al. Epidemiology of Castleman disease associated with AA amyloidosis: description of 2 new cases and literature review. Amyloid. 2019 Dec;26(4):197-202.
http://www.ncbi.nlm.nih.gov/pubmed/31364863?tool=bestpractice.com
[22]Bernabei L, Waxman A, Caponetti G, et al. AA amyloidosis associated with Castleman disease: a case report and review of the literature. Medicine (Baltimore). 2020 Feb;99(6):e18978.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7015640
http://www.ncbi.nlm.nih.gov/pubmed/32028407?tool=bestpractice.com
[23]Lachmann HJ. Periodic fever syndromes. Best Pract Res Clin Rheumatol. 2017 Aug;31(4):596-609.
http://www.ncbi.nlm.nih.gov/pubmed/29773275?tool=bestpractice.com
[24]Delaleu J, Deshayes S, Rodrigues F, et al. Tumour necrosis factor receptor-1 associated periodic syndrome (TRAPS)-related AA amyloidosis: a national case series and systematic review. Rheumatology (Oxford). 2021 Dec 1;60(12):5775-84.
https://academic.oup.com/rheumatology/article/60/12/5775/6170651
http://www.ncbi.nlm.nih.gov/pubmed/33715002?tool=bestpractice.com
[25]Rodrigues F, Cuisset L, Cador-Rousseau B, et al. AA amyloidosis complicating cryopyrin-associated periodic syndrome: a study of 86 cases including 23 French patients and systematic review. Rheumatology (Oxford). 2022 Nov 28;61(12):4827-34.
http://www.ncbi.nlm.nih.gov/pubmed/35262642?tool=bestpractice.com
[26]Lachmann HJ, Goodman HJ, Andrews PA, et al. AA amyloidosis complicating hyperimmunoglobulinemia D with periodic fever syndrome: a report of two cases. Arthritis Rheum. 2006 Jun;54(6):2010-4.
https://onlinelibrary.wiley.com/doi/epdf/10.1002/art.21901
http://www.ncbi.nlm.nih.gov/pubmed/16732551?tool=bestpractice.com
chronic inflammatory conditions (e.g., inflammatory polyarthropathy, inflammatory bowel disease [specifically Crohn disease])
chronic infections (e.g., bronchiectasis, tuberculosis, subcutaneous injection of illicit drugs, decubitus ulcers, chronic urinary tract infections, osteomyelitis)
familial periodic fever syndromes (e.g., familial Mediterranean fever, tumor necrosis factor [TNF] receptor-associated periodic fever syndromes [TRAPS], cryopyrin-associated periodic syndromes [CAPS; such as Muckle-Wells syndrome], mevalonate kinase deficiency [formerly known as hyper-IgD syndrome])
Castleman disease (plasma cell variant), a noncancerous lymphoproliferative disorder, but this is rare.
ATTRwt is associated with aging and affects mainly elderly men. History often includes cardiomyopathy, carpal tunnel syndrome, and spinal stenosis.[62]Law S, Gillmore JD. When to suspect and how to approach a diagnosis of amyloidosis. Am J Med. 2022 Apr;135 Suppl 1:S2-8.
https://www.amjmed.com/article/S0002-9343(22)00029-8/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/35081377?tool=bestpractice.com
Symptoms
Patients with amyloidosis frequently present with symptoms relating to a clinical syndrome of the affected organ (e.g., cardiomyopathy, nephrotic syndrome, neuropathy).[63]Palladini G, Milani P, Merlini G. Management of AL amyloidosis in 2020. Blood. 2020 Dec 3;136(23):2620-7.
https://www.sciencedirect.com/science/article/pii/S0006497120819582
http://www.ncbi.nlm.nih.gov/pubmed/33270858?tool=bestpractice.com
Fatigue, weight loss, paresthesias, and dyspnea on exertion are the most common symptoms associated with amyloidosis and are common to all systemic forms.[41]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis: diagnosis and management. Clin Lymphoma Myeloma. 2005 Nov;6(3):208-19.
http://www.ncbi.nlm.nih.gov/pubmed/16354326?tool=bestpractice.com
However, these complaints are nonspecific.
Extreme weight loss (e.g., >9 kg) is common (particularly in patients with cardiac and hepatic involvement) and is suggestive of amyloidosis if associated with edema or neuropathy.[48]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis. Best Pract Res Clin Haematol. 2005;18(4):709-27.
http://www.ncbi.nlm.nih.gov/pubmed/16026746?tool=bestpractice.com
Cardiac symptoms
Cardiac involvement is most commonly associated with AL amyloidosis and transthyretin (TTR)-related amyloidosis (hereditary and wild type).[41]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis: diagnosis and management. Clin Lymphoma Myeloma. 2005 Nov;6(3):208-19.
http://www.ncbi.nlm.nih.gov/pubmed/16354326?tool=bestpractice.com
[42]Shah KB, Inoue Y, Mehra MR. Amyloidosis and the heart: a comprehensive review. Arch Intern Med. 2006 Sep 25;166(17):1805-13.
http://archinte.jamanetwork.com/article.aspx?articleid=410996
http://www.ncbi.nlm.nih.gov/pubmed/17000935?tool=bestpractice.com
[48]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis. Best Pract Res Clin Haematol. 2005;18(4):709-27.
http://www.ncbi.nlm.nih.gov/pubmed/16026746?tool=bestpractice.com
[53]Siddiqi OK, Ruberg FL. Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med. 2018 Jan;28(1):10-21.
http://www.ncbi.nlm.nih.gov/pubmed/28739313?tool=bestpractice.com
[54]Kittleson MM, Maurer MS, Ambardekar AV, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020 Jul 7;142(1):e7-22.
https://www.doi.org/10.1161/CIR.0000000000000792
http://www.ncbi.nlm.nih.gov/pubmed/32476490?tool=bestpractice.com
[64]Muchtar E, Gertz MA, Kumar SK, et al. Improved outcomes for newly diagnosed AL amyloidosis between 2000 and 2014: cracking the glass ceiling of early death. Blood. 2017 Apr 13;129(15):2111-9.
https://www.doi.org/10.1182/blood-2016-11-751628
http://www.ncbi.nlm.nih.gov/pubmed/28126928?tool=bestpractice.com
Lightheadedness can be a symptom of cardiac amyloidosis (low cardiac output with preserved ejection fraction). Patients may have jaw claudication, calf and limb claudication, and rarely angina if there is involvement of the coronary arterioles.
Fatigue and dyspnea on exertion caused by early cardiac involvement are generally not recognized as symptoms of overt heart failure and can be misdiagnosed as being stress-related or functional.
Renal involvement
Most commonly associated with AL amyloidosis, AA amyloidosis, non-TTR hereditary amyloidosis (e.g., fibrinogen A alpha-chain, apolipoprotein A), and leukocyte chemotactic factor 2 (LECT2) amyloidosis.[3]Buxbaum JN, Dispenzieri A, Eisenberg DS, et al. Amyloid nomenclature 2022: update, novel proteins, and recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid. 2022 Dec;29(4):213-9.
https://www.tandfonline.com/doi/full/10.1080/13506129.2022.2147636
http://www.ncbi.nlm.nih.gov/pubmed/36420821?tool=bestpractice.com
[16]Lachmann HJ, Goodman HJB, Gilbertson JA, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007 Jun 7;356(23):2361-71.
https://www.nejm.org/doi/10.1056/NEJMoa070265
http://www.ncbi.nlm.nih.gov/pubmed/17554117?tool=bestpractice.com
[32]Rezk T, Gilbertson JA, Rowczenio D, et al. Diagnosis, pathogenesis and outcome in leucocyte chemotactic factor 2 (ALECT2) amyloidosis. Nephrol Dial Transplant. 2018 Feb 1;33(2):241-7.
https://academic.oup.com/ndt/article/33/2/241/2527565?login=false
http://www.ncbi.nlm.nih.gov/pubmed/29401357?tool=bestpractice.com
[39]Gertz MA, Leung N, Lacy MQ, et al. Clinical outcome of immunoglobulin light chain amyloidosis affecting the kidney. Nephrol Dial Transplant. 2009 Oct;24(10):3132-7.
https://www.doi.org/10.1093/ndt/gfp201
http://www.ncbi.nlm.nih.gov/pubmed/19403931?tool=bestpractice.com
[51]Cervantes CE, Atta MG. Kidney amyloidosis: updates on pathogenesis and therapeutic frontiers. Am J Nephrol. 2024 Jun 12:1-12.
https://karger.com/ajn/article/doi/10.1159/000539596/909119/Kidney-Amyloidosis-Updates-on-Pathogenesis-and
http://www.ncbi.nlm.nih.gov/pubmed/38865984?tool=bestpractice.com
[59]Papa R, Lachmann HJ. Secondary, AA, amyloidosis. Rheum Dis Clin North Am. 2018 Nov;44(4):585-603.
http://www.ncbi.nlm.nih.gov/pubmed/30274625?tool=bestpractice.com
Fatigue and lightheadedness are symptoms of nephrotic syndrome (hypoalbuminemia and intravascular volume contraction).
Neurologic symptoms
Nerve involvement can lead to peripheral and autonomic neuropathy, and is most commonly associated with AL amyloidosis and hereditary TTR (ATTRv) amyloidosis.[18]Vaxman I, Gertz M. When to suspect a diagnosis of amyloidosis. Acta Haematol. 2020;143(4):304-11.
https://www.doi.org/10.1159/000506617
http://www.ncbi.nlm.nih.gov/pubmed/32340017?tool=bestpractice.com
[48]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis. Best Pract Res Clin Haematol. 2005;18(4):709-27.
http://www.ncbi.nlm.nih.gov/pubmed/16026746?tool=bestpractice.com
[65]Conceição I, González-Duarte A, Obici L, et al. "Red-flag" symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016 Mar;21(1):5-9.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4788142
http://www.ncbi.nlm.nih.gov/pubmed/26663427?tool=bestpractice.com
[66]Planté-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011 Dec;10(12):1086-97.
http://www.ncbi.nlm.nih.gov/pubmed/22094129?tool=bestpractice.com
[67]Kaku M, Berk JL. Neuropathy associated with systemic amyloidosis. Semin Neurol. 2019 Oct;39(5):578-88.
http://www.ncbi.nlm.nih.gov/pubmed/31639841?tool=bestpractice.com
[68]Gonzalez-Duarte A, Valdés-Ferrer SI, Cantú-Brito C. Characteristics and natural history of autonomic involvement in hereditary ATTR amyloidosis: a systematic review. Clin Auton Res. 2019 Sep;29(suppl 1):1-9.
https://www.doi.org/10.1007/s10286-019-00630-y
http://www.ncbi.nlm.nih.gov/pubmed/31473866?tool=bestpractice.com
Mild peripheral neuropathy may occur in patients with ATTRwt amyloidosis (up to 20%).[55]Grogan M, Scott CG, Kyle RA, et al. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol. 2016 Sep 6;68(10):1014-20.
https://www.doi.org/10.1016/j.jacc.2016.06.033
http://www.ncbi.nlm.nih.gov/pubmed/27585505?tool=bestpractice.com
Nerve involvement is not a typical feature of AA amyloidosis, non-TTR hereditary amyloidosis, or LECT2 amyloidosis.[3]Buxbaum JN, Dispenzieri A, Eisenberg DS, et al. Amyloid nomenclature 2022: update, novel proteins, and recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid. 2022 Dec;29(4):213-9.
https://www.tandfonline.com/doi/full/10.1080/13506129.2022.2147636
http://www.ncbi.nlm.nih.gov/pubmed/36420821?tool=bestpractice.com
Initial presentation of peripheral neuropathy is usually distal symmetric sensory loss (i.e., loss of temperature and pain perceptions, followed by proprioceptive loss).[50]Kapoor M, Rossor AM, Jaunmuktane Z, et al. Diagnosis of amyloid neuropathy. Pract Neurol. 2019 Jun;19(3):250-8.
http://www.ncbi.nlm.nih.gov/pubmed/30598431?tool=bestpractice.com
Patients usually report dysesthesia and paresthesia of the feet and lower legs, which progress to the hands and arms over time.
Patients with autonomic neuropathy may have erectile dysfunction, orthostatic hypotension, gastrointestinal dysfunction, or urinary dysfunction.[18]Vaxman I, Gertz M. When to suspect a diagnosis of amyloidosis. Acta Haematol. 2020;143(4):304-11.
https://www.doi.org/10.1159/000506617
http://www.ncbi.nlm.nih.gov/pubmed/32340017?tool=bestpractice.com
[68]Gonzalez-Duarte A, Valdés-Ferrer SI, Cantú-Brito C. Characteristics and natural history of autonomic involvement in hereditary ATTR amyloidosis: a systematic review. Clin Auton Res. 2019 Sep;29(suppl 1):1-9.
https://www.doi.org/10.1007/s10286-019-00630-y
http://www.ncbi.nlm.nih.gov/pubmed/31473866?tool=bestpractice.com
Sweating abnormalities and failure of heart rate to change when body position is changed are signs of autonomic dysfunction.
Peripheral and autonomic neuropathy are important diagnostic clues for AL amyloidosis and ATTRv amyloidosis.[50]Kapoor M, Rossor AM, Jaunmuktane Z, et al. Diagnosis of amyloid neuropathy. Pract Neurol. 2019 Jun;19(3):250-8.
http://www.ncbi.nlm.nih.gov/pubmed/30598431?tool=bestpractice.com
Carpal tunnel syndrome prevalence is greatest in patients with TTR cardiac amyloidosis (20.3% vs. 4.1% in the general population), but it is also a manifestation of AL amyloidosis.[69]Milandri A, Farioli A, Gagliardi C, et al. Carpal tunnel syndrome in cardiac amyloidosis: implications for early diagnosis and prognostic role across the spectrum of aetiologies. Eur J Heart Fail. 2020 Mar;22(3):507-15.
https://onlinelibrary.wiley.com/doi/10.1002/ejhf.1742
http://www.ncbi.nlm.nih.gov/pubmed/31975495?tool=bestpractice.com
Tinel sign (tapping over the carpal nerve at the wrist produces tingling in the thumb, index, and middle finger) and Phalen maneuver (holding the dorsal surface of both hands together in forced flexion for around 1 minute produces tingling in the thumb, index, and middle finger) should be performed to test for carpal tunnel syndrome involvement in patients reporting paresthesia in the hands.
Gastrointestinal symptoms
Steatorrhea is typical of intestinal involvement. Severe fecal incontinence alternating with 3-4 days of constipation may be present.
Pseudo-obstructive symptoms (including nausea, vomiting, postprandial abdominal cramping) and gastroparesis may be present if there is upper gastrointestinal tract involvement.
Musculoskeletal symptoms
Musculoskeletal disorders (e.g., biceps tendon rupture, hip and knee osteoarthritis, trigger finger, and spinal stenosis) are typically associated with TTR amyloidosis, and may precede cardiac or neurologic manifestations.[70]Writing Committee, Kittleson MM, Ruberg FL, et al. 2023 ACC expert consensus decision pathway on comprehensive multidisciplinary care for the patient with cardiac amyloidosis: a report of the American College of Cardiology solution set oversight committee. J Am Coll Cardiol. 2023 Mar 21;81(11):1076-1126.
https://www.sciencedirect.com/science/article/pii/S073510972207423X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/36697326?tool=bestpractice.com
[71]Nativi-Nicolau JN, Karam C, Khella S, et al. Screening for ATTR amyloidosis in the clinic: overlapping disorders, misdiagnosis, and multiorgan awareness. Heart Fail Rev. 2022 May;27(3):785-93.
https://link.springer.com/article/10.1007/s10741-021-10080-2
http://www.ncbi.nlm.nih.gov/pubmed/33609196?tool=bestpractice.com
Physical exam
Common physical findings include lower extremity edema and elevated jugular venous distention (due to high right-sided filling pressure).[72]Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995 Jan;32(1):45-59.
http://www.ncbi.nlm.nih.gov/pubmed/7878478?tool=bestpractice.com
[73]Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation. 2005 Sep 27;112(13):2047-60.
https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.104.489187?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed
http://www.ncbi.nlm.nih.gov/pubmed/16186440?tool=bestpractice.com
Many physical findings of amyloidosis are specific and diagnostic for amyloidosis, but are present in ≤15% of patients.[48]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis. Best Pract Res Clin Haematol. 2005;18(4):709-27.
http://www.ncbi.nlm.nih.gov/pubmed/16026746?tool=bestpractice.com
Amyloid purpura: occurs in approximately 15% of patients with AL amyloidosis.[48]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis. Best Pract Res Clin Haematol. 2005;18(4):709-27.
http://www.ncbi.nlm.nih.gov/pubmed/16026746?tool=bestpractice.com
Typically periorbital but can occur anywhere above the nipple line.
Eyelid petechiae: common, but evident only when the patient's eyes are closed.
Macroglossia: specific and diagnostic for AL amyloidosis.[48]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis. Best Pract Res Clin Haematol. 2005;18(4):709-27.
http://www.ncbi.nlm.nih.gov/pubmed/16026746?tool=bestpractice.com
Occurs in approximately 10% of patients but is easily overlooked because the most common presentation is dental indentations on the underside of the tongue.
Enlargement of the submandibular salivary glands: specific for AL amyloidosis. It may be misinterpreted as lymphadenopathy. Salivary gland involvement results in a sicca syndrome. These patients are often misdiagnosed as having Sjögren syndrome.
Palpable hepatomegaly: >5 cm below the right costal margin, most commonly reported in AL amyloidosis (approximately 10% of patients).[16]Lachmann HJ, Goodman HJB, Gilbertson JA, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007 Jun 7;356(23):2361-71.
https://www.nejm.org/doi/10.1056/NEJMoa070265
http://www.ncbi.nlm.nih.gov/pubmed/17554117?tool=bestpractice.com
[48]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis. Best Pract Res Clin Haematol. 2005;18(4):709-27.
http://www.ncbi.nlm.nih.gov/pubmed/16026746?tool=bestpractice.com
Splenomegaly is usually of modest degree. Palpable hepatomegaly is uncommon in AA amyloidosis, but histopathologic changes in the liver may be evident. Liver involvement is rare in patients with hereditary amyloidosis.
Shoulder pad sign: rare, due to periarticular infiltration with amyloid; pseudohypertrophy is specific for AL amyloidosis.
Diffuse muscular weakness: amyloid myopathy can occur with muscle hypertrophy due to extracellular amyloid infiltration in the muscle, or can occur with muscular atrophy due to vascular occlusion leading to muscle ischemia and claudication.
Orthostatic hypotension with syncope: can occur if autonomic neuropathy is present (e.g., in AL amyloidosis or ATTRv amyloidosis).[Figure caption and citation for the preceding image starts]: Bilateral periorbital ecchymosis (amyloid purpura) in a patient with AL amyloidosisWilliams MU, Murphy CE, Gore RS, et al. BMJ Case Rep 2018;11:e225923. doi:10.1136/bcr-2018- 225923 [Citation ends].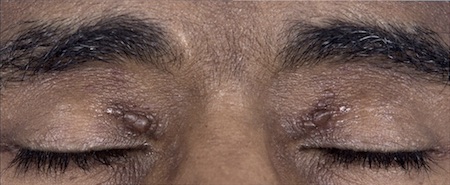 [Figure caption and citation for the preceding image starts]: Classic periorbital purpuraMorie A. Gertz, MD; courtesy of Mayo Clinic [Citation ends].
[Figure caption and citation for the preceding image starts]: Classic periorbital purpuraMorie A. Gertz, MD; courtesy of Mayo Clinic [Citation ends]. [Figure caption and citation for the preceding image starts]: Macroglossia in a patient with AL amyloidosisWilliams MU, Murphy CE, Gore RS, et al. BMJ Case Rep 2018;11:e225923. doi:10.1136/bcr-2018- 225923 [Citation ends].
[Figure caption and citation for the preceding image starts]: Macroglossia in a patient with AL amyloidosisWilliams MU, Murphy CE, Gore RS, et al. BMJ Case Rep 2018;11:e225923. doi:10.1136/bcr-2018- 225923 [Citation ends].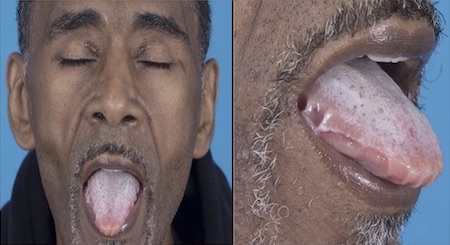
Key diagnostic tests
The first tests to order in patients with clinically suspected amyloidosis are immunofixation electrophoresis of the serum and urine (using 24-hour urine collection), and serum immunoglobulin free light chain assay.[61]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
[74]Dispenzieri A, Kyle R, Merlini G, et al. International Myeloma Working Group guidelines for serum-free light chain analysis in multiple myeloma and related disorders. Leukemia. 2009 Feb;23(2):215-24.
http://www.ncbi.nlm.nih.gov/pubmed/19020545?tool=bestpractice.com
Positive immunofixation (presence of a monoclonal protein in the serum or urine) and/or an abnormal serum immunoglobulin free light chain assay is reported in 99% of patients with AL amyloidosis.[75]Katzmann JA, Abraham RS, Dispenzieri A, et al. Diagnostic performance of quantitative kappa and lambda free light chain assays in clinical practice. Clin Chem. 2005 May;51(5):878-81.
https://www.doi.org/10.1373/clinchem.2004.046870
http://www.ncbi.nlm.nih.gov/pubmed/15774572?tool=bestpractice.com
A diagnosis of AL amyloidosis should be confirmed histologically (e.g., biopsy with amyloid typing) to avoid a misdiagnosis because an incidental monoclonal gammopathy may be present in other types of amyloidosis (e.g., hereditary amyloidosis, ATTRwt amyloidosis).[27]Comenzo RL, Zhou P, Fleisher M, et al. Seeking confidence in the diagnosis of systemic AL (Ig light-chain) amyloidosis: patients can have both monoclonal gammopathies and hereditary amyloid proteins. Blood. 2006 May 1;107(9):3489-91.
http://www.bloodjournal.org/content/107/9/3489.full
http://www.ncbi.nlm.nih.gov/pubmed/16439680?tool=bestpractice.com
[28]Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002 Jun 6;346(23):1786-91.
http://www.nejm.org/doi/full/10.1056/NEJMoa013354#t=article
http://www.ncbi.nlm.nih.gov/pubmed/12050338?tool=bestpractice.com
[60]Phull P, Sanchorawala V, Connors LH, et al. Monoclonal gammopathy of undetermined significance in systemic transthyretin amyloidosis (ATTR). Amyloid. 2018 Mar;25(1):62-7.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6157907
http://www.ncbi.nlm.nih.gov/pubmed/29424556?tool=bestpractice.com
[61]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
[76]Morris KL, Tate JR, Gill D, et al. Diagnostic and prognostic utility of the serum free light chain assay in patients with AL amyloidosis. Intern Med J. 2007 Jul;37(7):456-63.
http://www.ncbi.nlm.nih.gov/pubmed/17547724?tool=bestpractice.com
A diagnosis of AL amyloidosis is unlikely if immunofixation and serum immunoglobulin free light chain assay are normal. Patients with clinically suspected amyloidosis with equivocal or normal immunofixation and serum immunoglobulin free light chain assay should undergo a careful and prompt evaluation (including genetic testing) for other types of amyloidosis (e.g., AA amyloidosis, TTR amyloidosis, localized amyloidosis).
Biopsy studies
Histologic confirmation of amyloid deposits in tissue is essential for establishing a diagnosis of amyloidosis.
Bone marrow aspirate and biopsy, and subcutaneous fat aspirate (e.g., abdominal fat pad) are recommended in patients with suspected amyloidosis (e.g., if a monoclonal protein is present).[61]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
Other tissues that can be biopsied include lip (minor salivary gland) and rectum.[61]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
Bone marrow aspirate and biopsy can also be used to identify clonal plasma cells and assess for coexistent multiple myeloma. See Multiple myeloma.
If bone marrow and tissue biopsy studies are negative, biopsy of an involved organ (e.g., heart, liver, kidney, nerve) should be performed as clinically indicated.[61]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
Multiple tissue or organ biopsies are potentially hazardous and are not recommended.[77]Gillmore JD, Wechalekar A, Bird J, et al. Guidelines on the diagnosis and investigation of AL amyloidosis. Br J Haematol. 2015 Jan;168(2):207-18.
http://onlinelibrary.wiley.com/doi/10.1111/bjh.13156/full
http://www.ncbi.nlm.nih.gov/pubmed/25312307?tool=bestpractice.com
Bone marrow biopsy combined with subcutaneous fat aspirate (e.g., abdominal fat pad) will identify amyloid deposits in most (85%) patients with amyloidosis.[78]Muchtar E, Dispenzieri A, Lacy MQ, et al. Overuse of organ biopsies in immunoglobulin light chain amyloidosis (AL): the consequence of failure of early recognition. Ann Med. 2017 Nov;49(7):545-551.
https://www.doi.org/10.1080/07853890.2017.1304649
http://www.ncbi.nlm.nih.gov/pubmed/28271734?tool=bestpractice.com
Apple-green birefringence on a Congo red stained aspirate or biopsy specimen is required for diagnosis.[79]Gertz MA. The classification and typing of amyloid deposits. Am J Clin Pathol. 2004 Jun;121(6):787-9.
http://www.ncbi.nlm.nih.gov/pubmed/15198347?tool=bestpractice.com
Apple-green birefringence after Congo red staining confirms the presence of amyloid deposits but does not differentiate between different types of amyloid.[61]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
Amyloid typing and/or immunohistochemical studies should be carried out to confirm the type of amyloid.
Fluorescence in situ hybridization (FISH) studies should be done on bone marrow aspirate to identify molecular markers that can guide prognosis and treatment e.g., t(11;14) translocation.[80]Gertz MA, Dispenzieri A, Muchtar E. Importance of FISH genetics in light chain amyloidosis. Oncotarget. 2017 Oct 10;8(47):81735-6.
https://pmc.ncbi.nlm.nih.gov/articles/PMC5669844
[81]Muchtar E, Dispenzieri A, Kumar SK, et al. Interphase fluorescence in situ hybridization in untreated AL amyloidosis has an independent prognostic impact by abnormality type and treatment category. Leukemia. 2017 Jul;31(7):1562-9.
http://www.ncbi.nlm.nih.gov/pubmed/27904139?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Congo red stain blood vessel in a bone marrow biopsy demonstrating green birefringence pathognomonic of amyloidosisMorie A. Gertz, MD; courtesy of Mayo Clinic [Citation ends].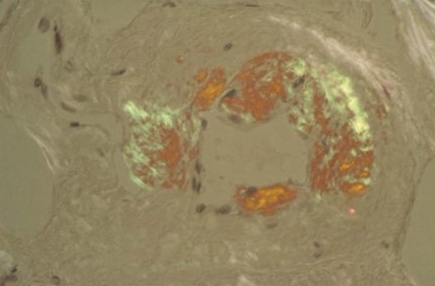
Amyloid typing
Mass spectrometry-based proteomic analysis is currently the gold standard for amyloid typing. It is the most direct method of confirming the amyloid type (e.g., light chain, serum amyloid A [SAA; associated with AA amyloidosis], TTR).
Immuno-electron microscopy can be used on renal biopsy specimens to clarify the fibrillar nature of the amyloid, but is not part of routine clinical practice for other biopsy material.[Figure caption and citation for the preceding image starts]: Electron micrograph demonstrating classical amyloid fibrilsMorie A. Gertz, MD; courtesy of Mayo Clinic [Citation ends].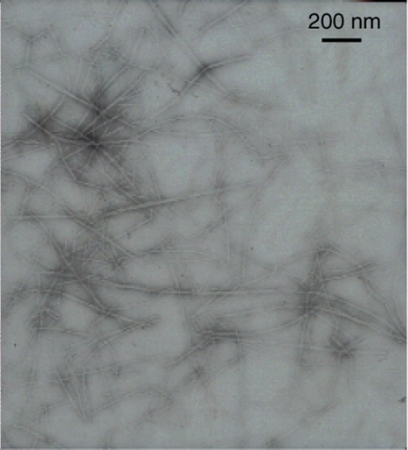
Immunohistochemical studies
Immunohistochemical staining of amyloid deposits can be attempted to distinguish the various forms of systemic amyloidosis. Commercially available antisera to immunoglobulin light chains, SAA, and TTR are typically used but may lack specificity and sensitivity.
Immunohistochemistry has lower diagnostic accuracy than mass spectrometry.[61]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
Genetic testing
Genetic testing can be used to assess for hereditary amyloidosis (e.g., ATTRv, fibrinogen A alpha-chain, apolipoprotein A, lysozyme) and familial periodic fever syndromes associated with AA amyloidosis (e.g., familial Mediterranean fever, TRAPS, CAPS [Muckle-Wells syndrome], mevalonate kinase deficiency [hyper-IgD syndrome]).[82]Gillmore JD, Reilly MM, Coats CJ, et al. Clinical and genetic evaluation of people with or at risk of hereditary ATTR amyloidosis: an expert opinion and consensus on best practice in Ireland and the UK. Adv Ther. 2022 Jun;39(6):2292-301.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9122857
http://www.ncbi.nlm.nih.gov/pubmed/35419651?tool=bestpractice.com
[83]Conceição I, Damy T, Romero M, et al. Early diagnosis of ATTR amyloidosis through targeted follow-up of identified carriers of TTR gene mutations. Amyloid. 2019 Mar;26(1):3-9.
https://www.tandfonline.com/doi/full/10.1080/13506129.2018.1556156
http://www.ncbi.nlm.nih.gov/pubmed/30793974?tool=bestpractice.com
Use of genetic testing is important to avoid a misdiagnosis (e.g., AL amyloidosis) in patients with hereditary amyloidosis.[29]Gertz M, Adams D, Ando Y, et al. Avoiding misdiagnosis: expert consensus recommendations for the suspicion and diagnosis of transthyretin amyloidosis for the general practitioner. BMC Fam Pract. 2020 Sep 23;21(1):198.
https://www.doi.org/10.1186/s12875-020-01252-4
http://www.ncbi.nlm.nih.gov/pubmed/32967612?tool=bestpractice.com
[61]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
Ancillary tests
Patients should have the following tests to guide diagnosis and prognosis, and to assess organ involvement:[61]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
CBC with differential
Peripheral blood smear, serum quantitative immunoglobulins, and serum protein electrophoresis, to assess for a plasma cell disorder
N‐terminal pro‐B-type natriuretic peptide (NT‐proBNP; B-type natriuretic peptide [BNP] if NT‐proBNP is unavailable), serum troponin T (troponin I if troponin T is unavailable), and lipid panel, to assess for heart involvement and for prognostication
Urine total protein and urine protein electrophoresis (from the 24-hour urine sample)
Comprehensive metabolic profile (including serum blood urea nitrogen [BUN], serum creatinine, electrolytes, serum albumin, serum calcium, serum uric acid, serum lactate dehydrogenase [LDH], beta-2 microglobulin; liver function tests [LFTs]), to assess for renal and liver involvement
Coagulation studies (including prothrombin time [PT], partial thromboplastin time [PTT], factor X), to assess for amyloid-related coagulation abnormalities
Orthostatic vital sign assessment and electromyogram (if clinically significant peripheral neuropathy is present)/nerve conduction studies, to assess for nerve involvement
Thyroid-stimulating hormone and cortisol levels, to assess for endocrine involvement
Pulmonary function tests, to assess for lung involvement
Cardiac evaluation
Cardiac involvement commonly occurs in AL, ATTRv, and ATTRwt amyloidosis.[41]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis: diagnosis and management. Clin Lymphoma Myeloma. 2005 Nov;6(3):208-19.
http://www.ncbi.nlm.nih.gov/pubmed/16354326?tool=bestpractice.com
[42]Shah KB, Inoue Y, Mehra MR. Amyloidosis and the heart: a comprehensive review. Arch Intern Med. 2006 Sep 25;166(17):1805-13.
http://archinte.jamanetwork.com/article.aspx?articleid=410996
http://www.ncbi.nlm.nih.gov/pubmed/17000935?tool=bestpractice.com
[48]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis. Best Pract Res Clin Haematol. 2005;18(4):709-27.
http://www.ncbi.nlm.nih.gov/pubmed/16026746?tool=bestpractice.com
[53]Siddiqi OK, Ruberg FL. Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med. 2018 Jan;28(1):10-21.
http://www.ncbi.nlm.nih.gov/pubmed/28739313?tool=bestpractice.com
[54]Kittleson MM, Maurer MS, Ambardekar AV, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020 Jul 7;142(1):e7-22.
https://www.doi.org/10.1161/CIR.0000000000000792
http://www.ncbi.nlm.nih.gov/pubmed/32476490?tool=bestpractice.com
[64]Muchtar E, Gertz MA, Kumar SK, et al. Improved outcomes for newly diagnosed AL amyloidosis between 2000 and 2014: cracking the glass ceiling of early death. Blood. 2017 Apr 13;129(15):2111-9.
https://www.doi.org/10.1182/blood-2016-11-751628
http://www.ncbi.nlm.nih.gov/pubmed/28126928?tool=bestpractice.com
Late diagnosis of cardiac involvement is associated with poor outcomes.[84]Tuzovic M, Yang EH, Baas AS, et al. Cardiac amyloidosis: diagnosis and treatment strategies. Curr Oncol Rep. 2017 Jul;19(7):46.
http://www.ncbi.nlm.nih.gov/pubmed/28528458?tool=bestpractice.com
An ECG and echocardiogram (with tissue Doppler and global longitudinal strain) should be carried out in all patients (symptomatic or asymptomatic) with suspected or confirmed cardiac amyloidosis.[54]Kittleson MM, Maurer MS, Ambardekar AV, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020 Jul 7;142(1):e7-22.
https://www.doi.org/10.1161/CIR.0000000000000792
http://www.ncbi.nlm.nih.gov/pubmed/32476490?tool=bestpractice.com
[70]Writing Committee, Kittleson MM, Ruberg FL, et al. 2023 ACC expert consensus decision pathway on comprehensive multidisciplinary care for the patient with cardiac amyloidosis: a report of the American College of Cardiology solution set oversight committee. J Am Coll Cardiol. 2023 Mar 21;81(11):1076-1126.
https://www.sciencedirect.com/science/article/pii/S073510972207423X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/36697326?tool=bestpractice.com
[85]Dorbala S, Ando Y, Bokhari S, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2-evidence base and standardized methods of imaging. Circ Cardiovasc Imaging. 2021 Jul;14(7):e000029.
https://www.ahajournals.org/doi/full/10.1161/HCI.0000000000000029?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org
http://www.ncbi.nlm.nih.gov/pubmed/34196223?tool=bestpractice.com
Increased left ventricular (LV) wall thickness, typical LV longitudinal strain pattern, and reduced tissue Doppler velocities are highly suggestive of cardiac amyloidosis.[54]Kittleson MM, Maurer MS, Ambardekar AV, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020 Jul 7;142(1):e7-22.
https://www.doi.org/10.1161/CIR.0000000000000792
http://www.ncbi.nlm.nih.gov/pubmed/32476490?tool=bestpractice.com
[85]Dorbala S, Ando Y, Bokhari S, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2-evidence base and standardized methods of imaging. Circ Cardiovasc Imaging. 2021 Jul;14(7):e000029.
https://www.ahajournals.org/doi/full/10.1161/HCI.0000000000000029?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org
http://www.ncbi.nlm.nih.gov/pubmed/34196223?tool=bestpractice.com
Cardiac MRI may be useful if an echocardiogram is suggestive or indeterminate. However, it is not diagnostic and cannot distinguish between AL and TTR cardiac amyloidosis.[70]Writing Committee, Kittleson MM, Ruberg FL, et al. 2023 ACC expert consensus decision pathway on comprehensive multidisciplinary care for the patient with cardiac amyloidosis: a report of the American College of Cardiology solution set oversight committee. J Am Coll Cardiol. 2023 Mar 21;81(11):1076-1126.
https://www.sciencedirect.com/science/article/pii/S073510972207423X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/36697326?tool=bestpractice.com
[85]Dorbala S, Ando Y, Bokhari S, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2-evidence base and standardized methods of imaging. Circ Cardiovasc Imaging. 2021 Jul;14(7):e000029.
https://www.ahajournals.org/doi/full/10.1161/HCI.0000000000000029?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org
http://www.ncbi.nlm.nih.gov/pubmed/34196223?tool=bestpractice.com
Cardiac MRI parameters should be combined with electrocardiographic, clinical, biomarker, and other imaging findings to maximize diagnostic accuracy.[85]Dorbala S, Ando Y, Bokhari S, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2-evidence base and standardized methods of imaging. Circ Cardiovasc Imaging. 2021 Jul;14(7):e000029.
https://www.ahajournals.org/doi/full/10.1161/HCI.0000000000000029?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org
http://www.ncbi.nlm.nih.gov/pubmed/34196223?tool=bestpractice.com
Cardiac scintigraphy with technetium-labeled bone tracers (99mTc-PYP or 99mTc-DPD) should be performed if TTR cardiac amyloidosis is suspected.[29]Gertz M, Adams D, Ando Y, et al. Avoiding misdiagnosis: expert consensus recommendations for the suspicion and diagnosis of transthyretin amyloidosis for the general practitioner. BMC Fam Pract. 2020 Sep 23;21(1):198.
https://www.doi.org/10.1186/s12875-020-01252-4
http://www.ncbi.nlm.nih.gov/pubmed/32967612?tool=bestpractice.com
Scintigraphy is sensitive for the detection of TTR cardiac amyloidosis and enables noninvasive clinical diagnosis.[70]Writing Committee, Kittleson MM, Ruberg FL, et al. 2023 ACC expert consensus decision pathway on comprehensive multidisciplinary care for the patient with cardiac amyloidosis: a report of the American College of Cardiology solution set oversight committee. J Am Coll Cardiol. 2023 Mar 21;81(11):1076-1126.
https://www.sciencedirect.com/science/article/pii/S073510972207423X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/36697326?tool=bestpractice.com
[85]Dorbala S, Ando Y, Bokhari S, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2-evidence base and standardized methods of imaging. Circ Cardiovasc Imaging. 2021 Jul;14(7):e000029.
https://www.ahajournals.org/doi/full/10.1161/HCI.0000000000000029?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org
http://www.ncbi.nlm.nih.gov/pubmed/34196223?tool=bestpractice.com
[86]Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016 Jun 14;133(24):2404-12.
https://www.doi.org/10.1161/CIRCULATIONAHA.116.021612
http://www.ncbi.nlm.nih.gov/pubmed/27143678?tool=bestpractice.com
[87]Castano A, Haq M, Narotsky DL, et al. Multicenter study of planar technetium 99m pyrophosphate cardiac imaging: predicting survival for patients with ATTR cardiac amyloidosis. JAMA Cardiol. 2016 Nov 1;1(8):880-9.
https://www.doi.org/10.1001/jamacardio.2016.2839
http://www.ncbi.nlm.nih.gov/pubmed/27557400?tool=bestpractice.com
However, it lacks specificity, therefore false positives may be seen. A negative monoclonal protein result alongside scintigraphy cardiac uptake (grade 2 or 3) confirms a diagnosis of TTR amyloidosis (without requiring biopsy).[61]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
[70]Writing Committee, Kittleson MM, Ruberg FL, et al. 2023 ACC expert consensus decision pathway on comprehensive multidisciplinary care for the patient with cardiac amyloidosis: a report of the American College of Cardiology solution set oversight committee. J Am Coll Cardiol. 2023 Mar 21;81(11):1076-1126.
https://www.sciencedirect.com/science/article/pii/S073510972207423X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/36697326?tool=bestpractice.com
If scintigraphy cardiac uptake is 1 or 0, a biopsy is required (cardiac or non-cardiac, depending on presentation).[61]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
In older men with an echocardiogram consistent with cardiac amyloidosis, a cardiac scintigraphy showing uptake of 99mTc-PYP or 99mTc-DPD in myocardial tissue increases suspicion for ATTRwt amyloidosis.
Cardiac workup for coronary artery disease is invariably normal.[88]Neben-Wittich MA, Wittich CM, Mueller PS, et al. Obstructive intramural coronary amyloidosis and myocardial ischemia are common in primary amyloidosis. Am J Med. 2005 Nov;118(11):1287.
http://www.ncbi.nlm.nih.gov/pubmed/16271914?tool=bestpractice.com
Imaging studies
Whole-body low-dose computed tomography (CT) or 18F-fluorodeoxyglucose positron emission tomography (FDG-PET)/CT can be used to detect osteolytic bone lesions if a monoclonal protein is detected. A skeletal survey can be used if these advanced imaging modalites are unavailable, but sensitivity is significantly lower.[61]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
A chest CT can be used to evaluate lung involvement, if clinically indicated.[61]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
Gastric emptying scan (if gastroparesis is present) and abdominal ultrasound or CT (as clinically indicated) can be used to evaluate liver and gastrointestinal involvement.[61]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
Upper and lower endoscopies can be performed if symptoms suggest gastrointestinal involvement.
123I-labeled serum amyloid P (SAP) scintigraphy can be used to assess the extent of organ involvement and dysfunction at diagnosis and during follow-up. SAP scintigraphy is standard practice in the UK and the Netherlands, but use in other countries may vary.[77]Gillmore JD, Wechalekar A, Bird J, et al. Guidelines on the diagnosis and investigation of AL amyloidosis. Br J Haematol. 2015 Jan;168(2):207-18.
http://onlinelibrary.wiley.com/doi/10.1111/bjh.13156/full
http://www.ncbi.nlm.nih.gov/pubmed/25312307?tool=bestpractice.com
[89]Hawkins PN, Lavender JP, Pepys MB. Evaluation of systemic amyloidosis by scintigraphy with 123I-labeled serum amyloid P component. N Engl J Med. 1990 Aug 23;323(8):508-13.
https://www.doi.org/10.1056/NEJM199008233230803
http://www.ncbi.nlm.nih.gov/pubmed/2377176?tool=bestpractice.com
Prognostication
Prognostic biomarkers for amyloidosis include: serum troponin T (troponin I if troponin T is unavailable); NT‐proBNP (BNP if NT-proBNP is unavailable); and the difference between involved and uninvolved serum free light chains (dFLC).
Serum troponin level is a sensitive marker for myocardial injury in amyloidosis; NT‐proBNP is a sensitive marker for myocardial stretch and congestive heart failure.[90]Dispenzieri A, Kyle RA, Gertz MA, et al. Survival in patients with primary systemic amyloidosis and raised serum cardiac troponins. Lancet. 2003 May 24;361(9371):1787-9.
http://www.ncbi.nlm.nih.gov/pubmed/12781539?tool=bestpractice.com
[91]Palladini G, Campana C, Klersy C, et al. Serum N-terminal pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation. 2003 May 20;107(19):2440-5.
https://www.ahajournals.org/doi/abs/10.1161/01.CIR.0000068314.02595.B2
http://www.ncbi.nlm.nih.gov/pubmed/12719281?tool=bestpractice.com
Troponin, NT‐proBNP, and dFLC are used in the Mayo staging criteria for AL amyloidosis.[92]Dispenzieri A, Gertz MA, Kyle RA, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol. 2004 Sep 15;22(18):3751-7.
http://www.ncbi.nlm.nih.gov/pubmed/15365071?tool=bestpractice.com
[93]Kumar S, Dispenzieri A, Lacy MQ, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012 Mar 20;30(9):989-95.
http://www.ncbi.nlm.nih.gov/pubmed/22331953?tool=bestpractice.com
[94]Wechalekar AD, Schonland SO, Kastritis E, et al. A European collaborative study of treatment outcomes in 346 patients with cardiac stage III AL amyloidosis. Blood. 2013 Apr 25;121(17):3420-7.
https://www.doi.org/10.1182/blood-2012-12-473066
http://www.ncbi.nlm.nih.gov/pubmed/23479568?tool=bestpractice.com
See Criteria.
Beta-2-microglobulin is predictive of survival in patients with AL amyloidosis.[95]Zerbini CA, Anderson JJ, Kane KA, et al. Beta 2 microglobulin serum levels and prediction of survival in AL amyloidosis. Amyloid. 2002 Dec;9(4):242-6.
http://www.ncbi.nlm.nih.gov/pubmed/12557752?tool=bestpractice.com
[96]Al Saleh AS, Sidiqi MH, Muchtar E, et al. Prognostic role of beta-2 microglobulin in patients with light chain amyloidosis treated with autologous stem cell transplantation. Biol Blood Marrow Transplant. 2020 Aug;26(8):1402-1405.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7371505
http://www.ncbi.nlm.nih.gov/pubmed/32422250?tool=bestpractice.com